Reagents and enzymes
The LwaCas13a enzyme was obtained from GenScript and stored at -80 °C in 50 mM Tris–HCl, 600 mM NaCl, 5% glycerol, and 2 mM DTT, pH 7.5. PAGE Ultramer DNA oligos for RNA guide synthesis were obtained from Integrated DNA Technologies (IDT, United States). A HiScribe™ T7 Quick High Yield RNA Synthesis Kit containing T7 polymerase, RNase inhibitor, and NTP mix buffer was obtained from New England Biolabs (NEB, United States). PCR primers were obtained from Eurogentec (Belgium). Hydroxyethyl piperazine ethane sulfonic acid (HEPES) and dimethylsulfoxide (DMSO) were obtained from Sigma‒Aldrich (United States).
Cell culture
BxPC-3, AsPC-1, and MIA PaCa-2 cells were maintained in Dulbecco’s minimal essential medium (DMEM, Invitrogen, Saint Aubin, France), and Capan-1 cells were maintained in Roswell Park Memorial Institute (RPMI, Invitrogen). For both media, 10% fetal bovine serum (FBS, Invitrogen), 100 U/mL penicillin (Invitrogen), and 100 μg/mL streptomycin (Invitrogen) were added. All cell lines were cultured at 37 °C and 5% CO2 in a humidified chamber.
Patient inclusion and sample collection
Patients were recruited prospectively between February 2023 and March 2023. All patients who received endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) in the context of a pancreatic mass during these 2 months were recruited. The patients’ demographic information is summarized in Table 1.
Table 1 Patient demographicsDNA extraction and quantification
DNA samples for KRAS detection were extracted from pancreatic tumor cell lines using a QIAamp DNA Extraction Kit® (Qiagen, France). KRASWT/WT DNA and KRASG12D/G12D, KRASG12C/G12C, and KRASG12V/G12V mutant DNA were extracted from the BxPC-3, AsPC-1, MIA PaCa-2, and Capan-1 cell lines, respectively, and verified by NGS analysis using the Bordeaux University Hospital Tumor Biology Department routine solid tumor panel (custom AmpliSeq panel with an Ion Torrent S5 sequencer (Thermo Fisher Scientific, United States)). All DNA samples were quantified by spectrophotometry using a Nanodrop® One/One device (Thermo Fisher Scientific, United States). For molecular analysis of the needle-rinsing fluids, a Maxwell RSC ccfDNA Plasma Kit was used for DNA extraction, and a DS11FX automated system (DeNovix) was used for concentration evaluation. The patient sample DNA concentrations are reported in Supplemental Table 1.
RNA guide synthesis and purification
Guide RNAs were produced by T7-mediated in vitro transcription as described by Kellner et al. [11]. Briefly, oligonucleotides (PAGE Ultramer DNA oligos from Integrated DNA Technologies) were resuspended at a concentration of 100 µM. Annealing was performed at 95 °C for 5 min, followed by a slow temperature decrease to 4 °C (0.1 °C/s) using common forward p.T7 oligo and Taq buffer (10X). In vitro transcription was next performed overnight with the HiScribe™ T7 Quick High Yield RNA Synthesis Kit (NEB, MA, USA) following the manufacturer’s instructions, and the products were subsequently purified with Agencourt RNAClean XP beads (Beckman Coulter). Purified RNA products were aliquoted and frozen at -80 °C.
DNA amplification step
PCR and allele-specific PCR amplification were performed using Phire Tissue Direct PCR Master Mix® (Thermo Fisher Scientific) following the manufacturer's instructions. The amplification primers and related annealing temperatures used are listed in Supplemental Table 2. All amplifications were performed using 10 ng of gDNA input, except for patient samples with insufficient DNA concentrations (Supplemental Table 1). An overhang including the T7 promoter was used to enable subsequent T7-mediated in vitro transcription of the PCR products [11]. All primers were used at a concentration of 250 nM. By default, 35 cycles of amplification were performed. For CASPER, after multiple conditions were tested, only 30 cycles of amplification were performed to optimize specificity.
CRISPR-Cas13a detection step
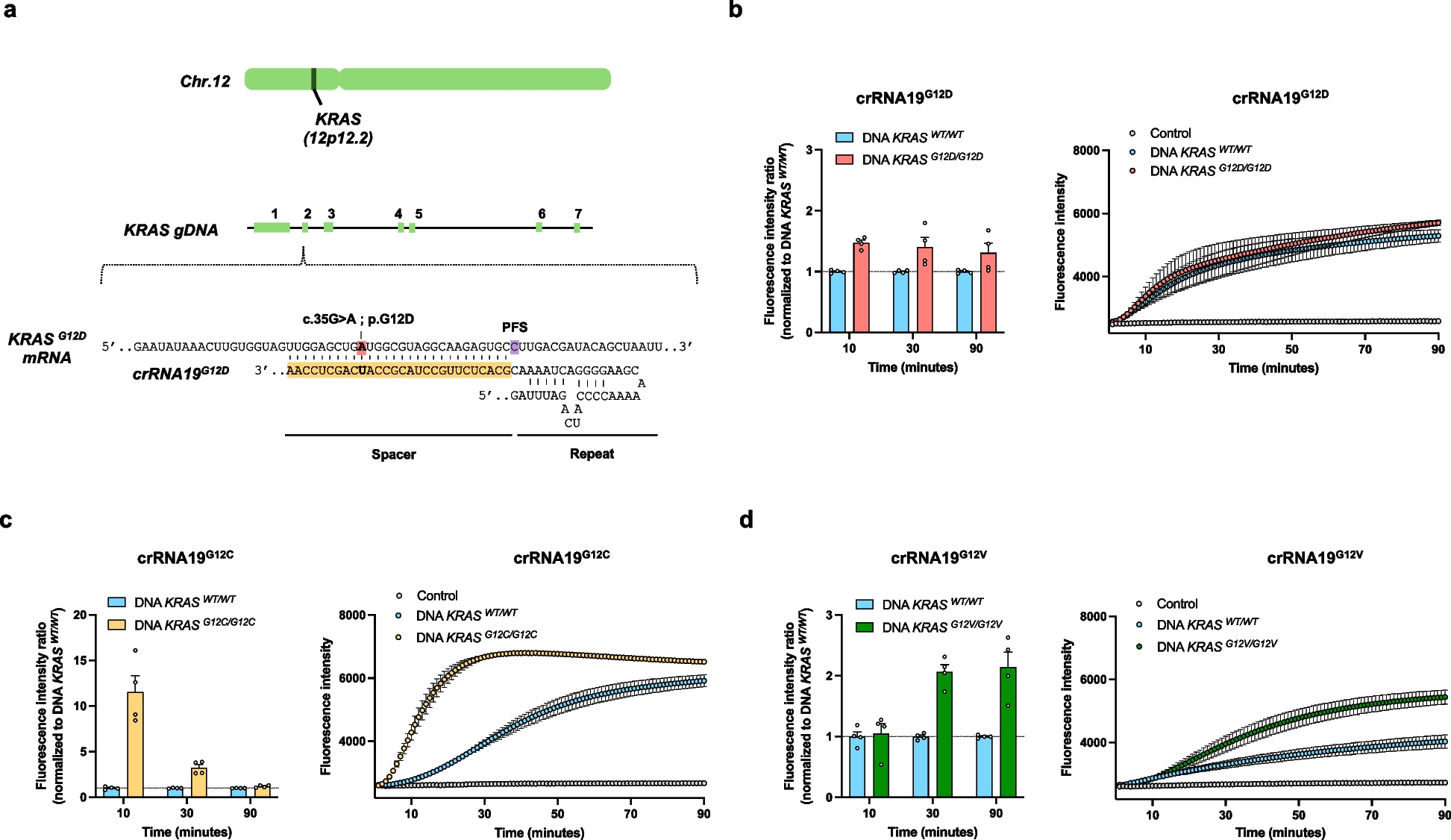
The RNA guide spacer sequences used are listed in Supplemental Table 2 and Supplemental Figs. 1 to 5. In vitro transcription of KRAS PCR products and Cas13-mediated detection of T7-produced RNA were performed simultaneously as described previously [11]. The detection mixture included 16 mM HEPES, 7.2 mM MgCl2, 640 nM rNTP, 0.05 U/µL T7 RNA polymerase, 1.6 × 103 U/µL murine RNase inhibitor (NEB), 5 µg/µL LwaCas13a protein, 400 pg/µL RNA guide, and 100 nM fluorescent RNA reporter. The final volume of the reaction was 20 µl, which included 1 µL of PCR products. All manipulations were performed on ice. After the addition of PCR products, the samples were immediately transferred to a CFX96 Touch Real-Time PCR Detection System (Bio-Rad), and the fluorescence level was quantified every minute for 90 min. Analysis of the results was performed using CFX MaestroTM software (Bio-Rad). The fluorescence intensity ratio was calculated at 90 min as follows:
Real-time quantitative PCR
The detection of the KRASG12D and KRASWT alleles was performed using a Promega GoTaq® qPCR kit (Promega, Wisconsin) following the manufacturer's instructions with 10 ng of DNA input. The primers used and annealing temperatures are summarized in Supplemental Table 2. All primers were used at a concentration of 2.5 µM. By default, 35 cycles of amplification were performed. The data were analyzed with CFX Maestro Software (Bio-Rad). The relative expression of the KRASG12D and KRASWT alleles was first normalized to that of GAPDH and then represented as fold changes (2−ΔΔCt). Melting curves showed that primers amplified only the specific fragments.
Droplet digital PCR
Droplet digital PCR analyses were performed on the Bio-Rad ddPCR platform (Bio-Rad, United States) with a QX-200 TM droplet generator and a QX-200 TM droplet reader. Bio-Rad KRAS G12/G13 screening and Bio-Rad KRAS G12D-specific kits were used for global or specific KRASG12 mutant detection according to the manufacturer's instructions. To compare the performance of ddPCR vs. PCR-CRISPR-Cas13a or CASPER, all experiments were performed using 10 ng of DNA. For patient samples, various amounts of DNA (18 µL, regardless of DNA concentration) were used for the KRAS G12/13 multiplex ddPCR screening assay, and 10 ng was used for the KRAS G12D-specific ddPCR assay, except for patient samples with insufficient DNA concentrations (Supplemental Table 1). Analysis of the results was performed using QuantaSoftTM software (Bio-Rad) with laboratory-validated clinical routine interpretation guidelines. For each assay, a wild-type sample and a “no DNA input” control were analyzed. The MAF positivity threshold (0.056%) was previously determined [12]. A sample was considered positive when the lower standard deviation value of the MAF was greater than the positivity threshold.
RNA secondary structure analysis
The predicted secondary RNA structures of the KRAS T7 RNA products were obtained with RNAfold® software (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). The default parameters were used.
Statistical analysis
Statistical tests were performed using Graph-Pad Prism software (v6.04). The results are expressed as the mean ± SEM or mean ± SD and were analyzed by unpaired, bilateral Student's t tests with Welch’s correction. Correlation analyses were performed using Spearman’s test. p < 0.05 was considered to indicate statistical significance.






留言 (0)