
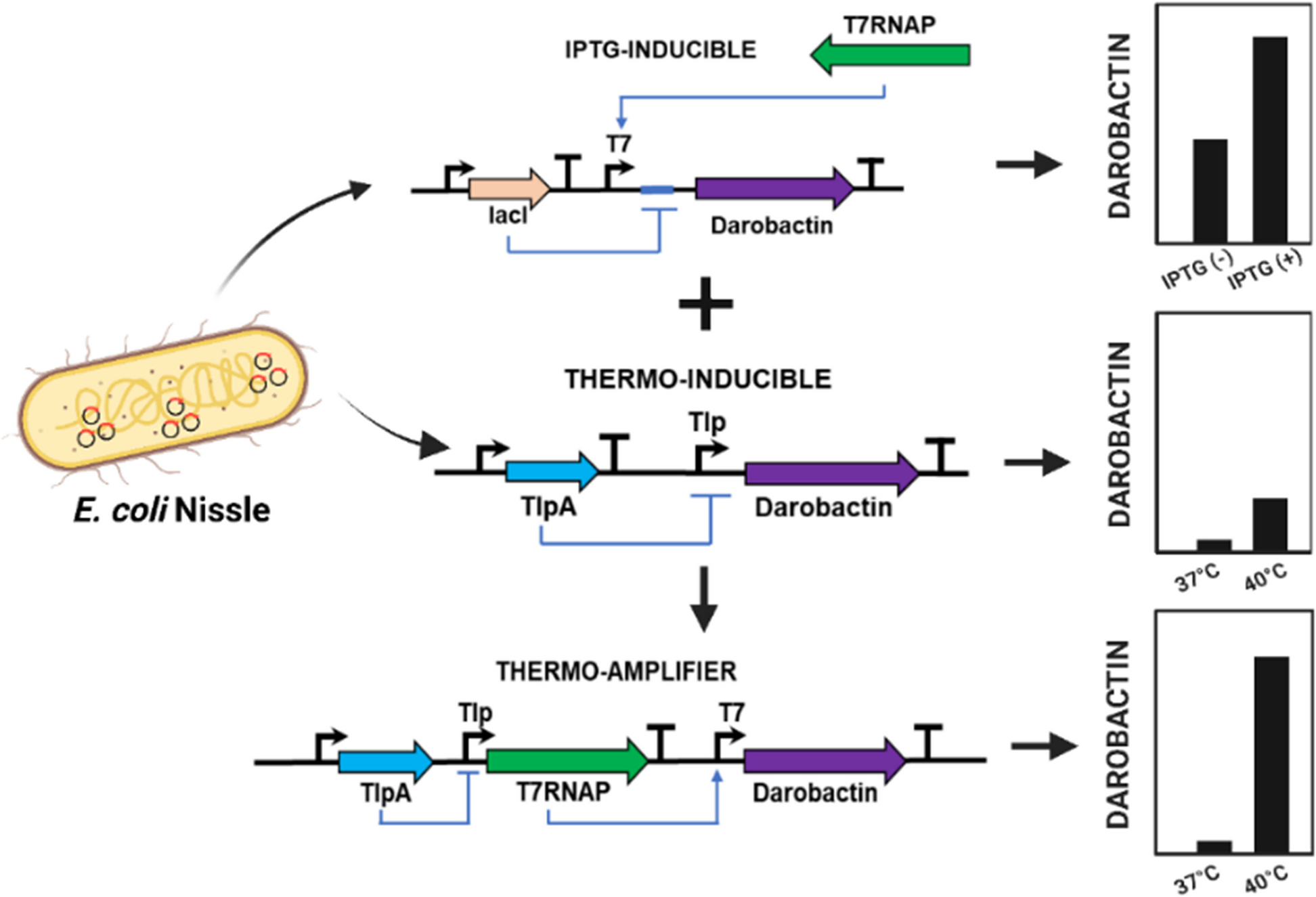
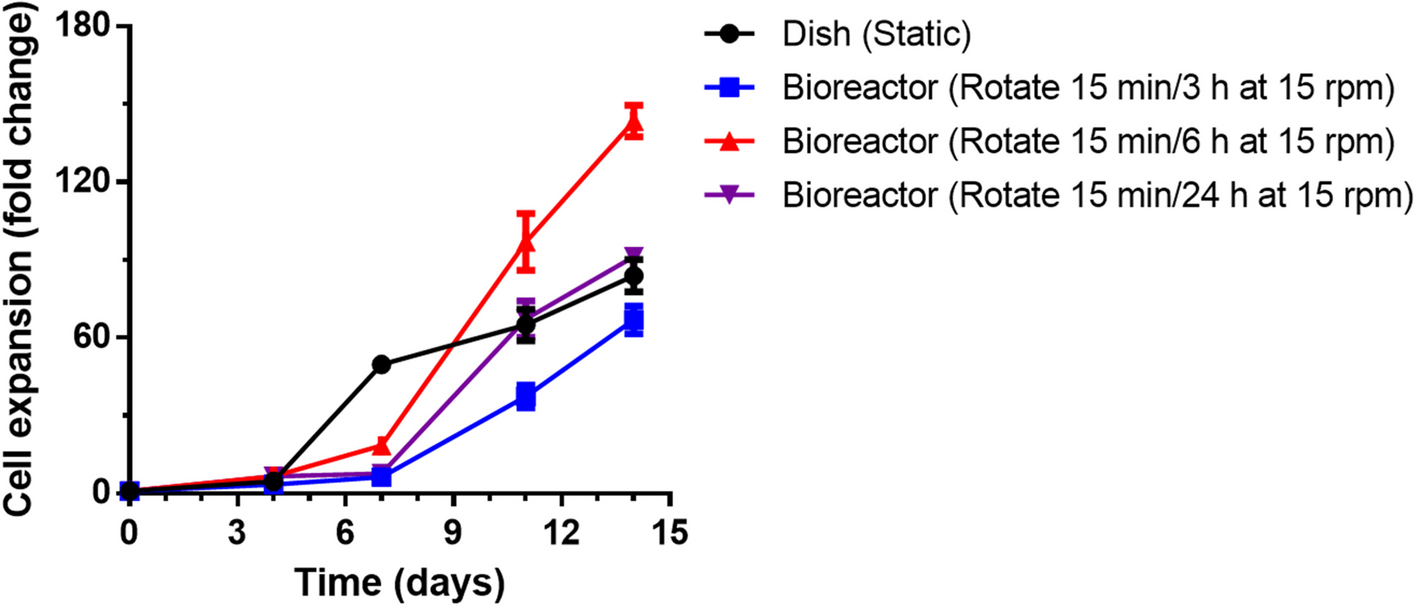
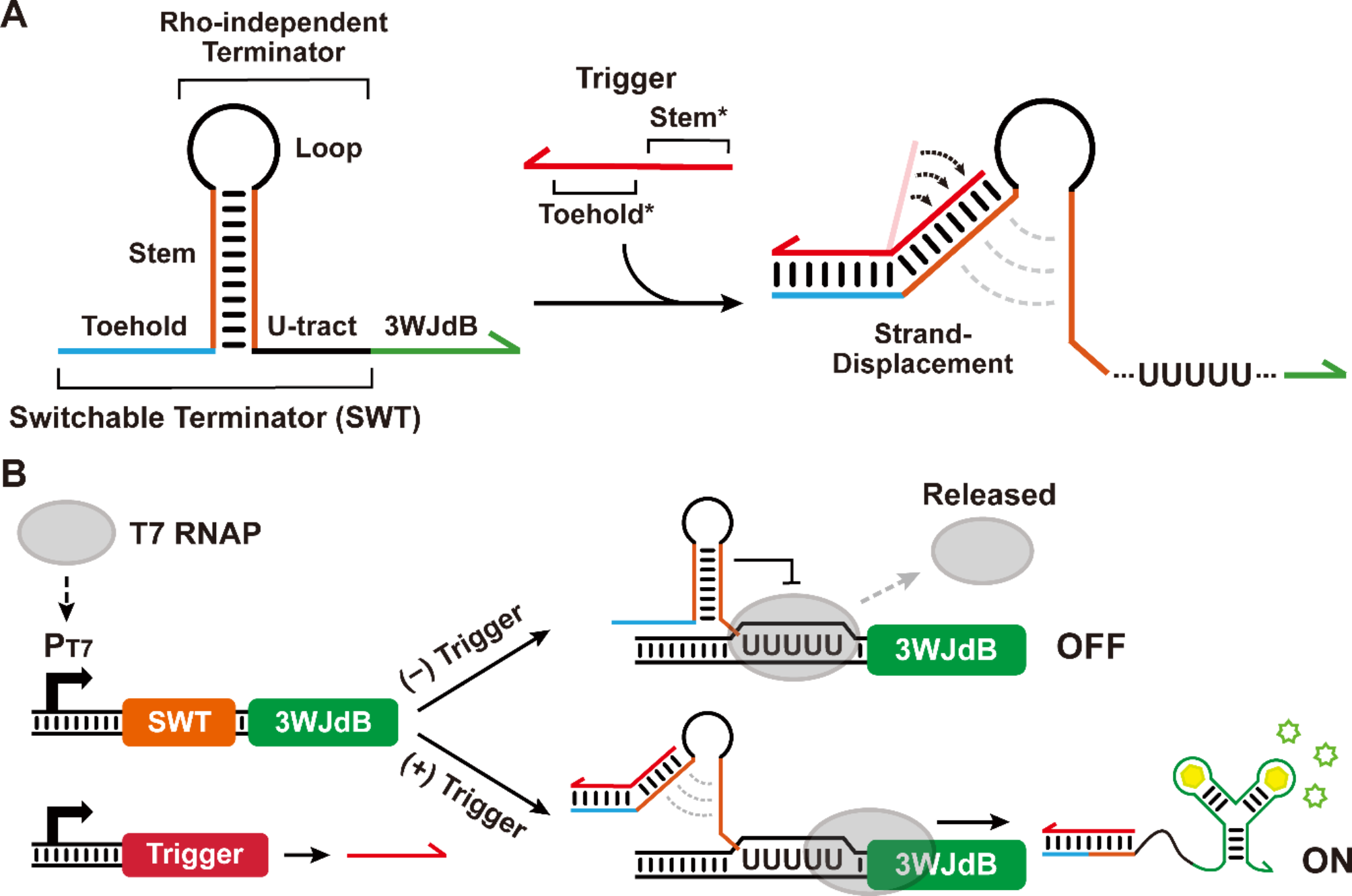
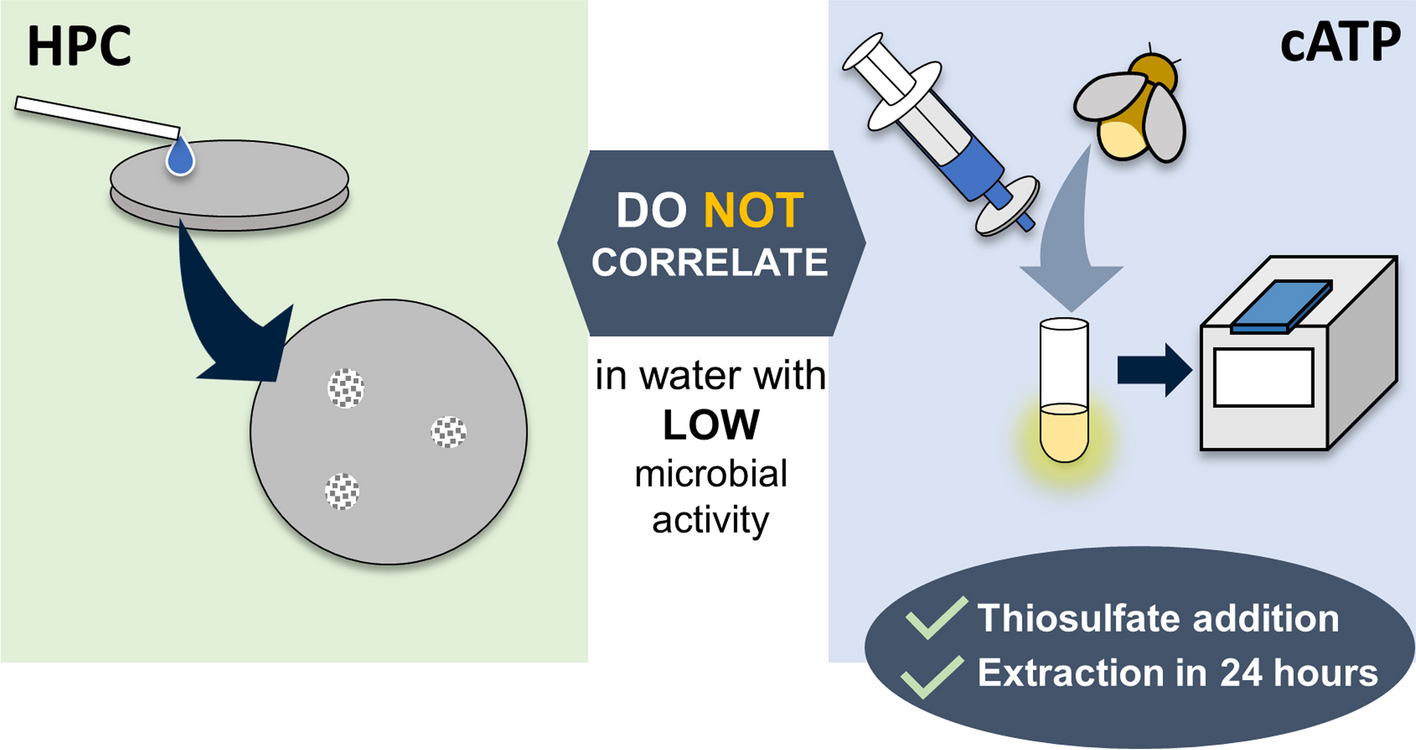
記住我






The structures of Chitosan and Eudragit used for MD simulations are shown in Fig. 1a. Results from both AA-MD and CG-MD simulations have been compared with experimental data for initial evaluations. Figure 1b exhibits PAX release (%) from computational approaches with experimental results. Interestingly, computational and experimental results are in appropriate agreement. In a comprehensive view, both mathematical approaches can catch the trend in the release diagram vs. pH conditions and anticipate comparable data to the reported real numbers. AA-MD results are lower than experimental data in all pH conditions, while CG-MD predicts more than experimental release. However, considering the trend of release values, the differences between computed and reported data are negligible.
Fig. 1
Initial comparison of the computational approaches with reported real data of PAX-Citosan-Eudragit. a molecular structure of Chitosan and Eudragit used in simulations. b Drug release (%) in varying pH conditions for real and mathematical methods (AA-MD and CG-MD). c i,ii) Comparison between hydrodynamic radius (Rh) of NPs (with different reaction times) with radius of gyration (Rg) (during simulations) obtained from computational methods at pH = 5 and 7, respectively. All diagrams are in good agreement with eachother, i.e., simulations anticipate the variation of Rg similar to the size of NPs. Experimental data was obtained from [44]
Furthermore, to validate computational results with reported experimental data, the formation of NPs through hydrodynamic radius (Rh) and the radius of gyration (Rg) obtained through simulations were compared (Fig. 1c). The Rg resembles the self-assembly of polymeric chains in the lab condition throughout the simulation. Moreover, Rg can be interpreted as the stability of the NPs’ model, i.e., the less the Rg size indicates the tighter and more stable particle. In this line, the diagrams showing the Rg change over the simulation time (in nanosecond scale) were prepared to be compared with the hydrodynamic size of NPs in the lab that has been prepared in the various reaction periods (based on hour scale). The change in Rg vs. AA-MD and CG-MD simulation times resembles the change in the size of actual NPs vs. reaction times. Altogether, the agreements between Rg and Rh, in addition to the release results, provide other pieces of evidence on the reliability of the computational procedures and approaches to continue for further investigations.
The interaction of the Chitosan molecule with the PAX molecule is simulated at different pHs. Gyration radius and energy analysis, charge distribution, the RDF and SASA diagrams are supposed to illustrate the results of this computational study. These results are obtained from both AA-MD and CG-MD simulations. The results of a comparison of analyses show PAX adsorption and release at specific pH levels.
Validation of simulations with experimental resultsFigure 2 shows the release of PAX employing nanocarrier via simulation methods of AA-MD and CG-MD. The simulation results show a similar release percentage with the results of the experimental work. Experimental data was obtained from a previously published research by Hasani-Sadrabadi et al. [44], in which they assessed various pH conditions on PAX delivery through multiple nanocarriers. Consistency of the simulation and experimental work results demonstrates the validity of the method and algorithm used in this work.
Fig. 2
The comparison of PAX release (%) by nanocarrier through (a) AA-MD, and (b) CG-MD simulation methods. Experimental data was obtained from [44]
Figure 3 illustrates the changes in nanocarrier size during simulation by AA-MD and CG-MD methods in blue and purple, respectively. Also, the nanocarrier size of the experimental work is shown in red. The process of altering nanocarrier size in computational and experimental work is similar. This result shows the accuracy of the methods and algorithms used in this work.
Fig. 3
The comparison among nanocarrier size resulted from computational and experimental works
Table 1 shows the average energy obtained from the simulation in the canonical (NVT) ensemble stage, and the experimental work. The average energy similarity in the NVT phase shows the accuracy of the methods and algorithms used in this work. At the NVT stage, the number of moles, temperature, and volume of the simulation box are fixed during the simulation. Molecular simulations are very sensitive to the force-field and potential functions. The potential function that the reference simulation used is different from the potential function of all-atom simulation. This caused a difference in the results as shown in Table 1. In coarse-grained simulations, this difference is much less (less than one percent). Given that molecular simulations have many limitations in terms of dimensions, potential functions, assumptions, etc., more in qualitative studies and are semi-quantitative, so the differences are usual in molecular simulations as long as they do not cause conflict in the trends and the qualitative understanding of the subjects.
Table 1 The average energy obtained from the simulation in the NVT stage and the experimental situation. Experimental data (reference simulation) was obtained from [44]AA-MD simulationIn AA-MD simulation, the results are presented on smaller scales but more accurately. The first analysis is the charge distribution analysis, which shows the charge density along with the simulation box (with Coulomb/box volume unit) and is made for PAX molecules, Chitosan, and Eudragit monomers (Fig. 4). Figure 4a shows the charge distribution for the Eudragit monomer. In this monomer, positive–negative charges are seen in proportion, considering that the ratio of negative charges is more than positive charges, and the charge of this polymer is negative. Also, as shown in Fig. 4b, Chitosan monomer has different charge distributions. In this monomer, there are positive and negative charges, where the positive charge is due to the presence of the amine group, and the negative charge is linked to the presence of the OH group. According to Fig. 4c, the PAX molecule has a relatively negative charge, and this is due to the OH group being in its structure. Also, the binding of PAX and nanocarrier after simulation is shown in Fig. 4d.
Fig. 4
Charge distribution in the structure of molecules (a) Eudragit, (b) Chitosan, (c) PAX, and (d) Binding of PAX molecules and nanocarrier after simulation
Figure 5 also shows the energy diagrams and the number of hydrogen bonds for the PAX molecule and the Eudragit and Chitosan polymers at different pHs. The energy diagram is calculated based on the electrostatic van der Waals energy and the total energy. The van der Waals energy results from the interaction among the molecules, based on the Leonard Jones equation [45]. Regarding this equation and based on the various molecules and atoms' mass, van der Walls energy calculations are obtained. Electrostatic energy is also calculated depending on each atom’s charge, derived from Coulomb's law [46]. In AA-MD simulation, each atom has charge and mass and based on these two parameters, van der Waals and electrostatic energies are calculated. The total energy is obtained regarding the sum of van der Waals and electrostatic energies. Energy analysis is essential in pH-dependent simulations since with the change of pH, both the charge of the atoms and the number of hydrogens and proteins in the system change. The charge alter causes immediate changes in electrostatic and total energy and consequently affects van der Waals energy.
Fig. 5
Energy diagram of the interactions among PAX and nanocarrier over time at (a) pH = 5; and (b) pH = 7; (c) hydrogen bonds formed between PAX and nanocarrier at pH = 5 and pH = 7
The diagrams in Fig. 5 demonstrate that with the acidification of the system, the number of protons has also increased; hence its electrostatic energy has become positive. At the pH = 7 (neutral), the average total energy is approximately -800, while at the pH = 5, the number of protons increases, and the charge becomes positive, and the average total energy is about 1500. However, van der Waals energy, which depends on atoms mass, has changed very quitely. These results indicate that at neutral pH, PAX adsorption occurred through Chitosan and Eudragit nanocarriers, while at the acidic pH, PAX excretion would occur. Therefore, it could be concluded that the PAX excretion also takes place at the acidic pH of the cancerous tumor. Another analysis that is essential in pH-dependent simulations is the analysis of hydrogen bonds, which are more potent than van der Waals and electrostatic energies. pH changes are evident in Fig. 5c as the number of hydrogen bonds increases in terms of simulation time. Also, protonation depends on pH changes.
The number of hydrogen bonds established at neutral pH is greater than the number of hydrogen bonds established at acidic pH. According to the Arrhenius theory [47], the lower average of hydrogen bonding at acidic pH indicates that nanocarrier and drug molecules release themselves in the hydrogen environment in cancer cells’ acidic conditions. This phenomenon resulted in positive electrostatic energy in Fig. 5a and the number of hydrogen bonds in Fig. 5c. Thus, at acidic pH, not only do the molecules gain the same charge, but also, they lose their hydrogen to form a hydrogen bond, resulting in the excretion of PAX molecules from the nanocarrier. Figure 5c shows that the average hydrogen bond increases slightly by 40 ns. This point indicates that from 40 ns onwards, the adsorption of PAX molecules into the nanocarrier increases and the drug bind to the nanocarrier.
Figure 6 shows the radial distribution function (RDF) diagram and the gyration radius. The RDF diagram depicts the distribution of PAX loads around the nanocarrier at different pHs. The maximum of this graph shows the highest adsorption rate among PAX and nanocarrier molecules. As shown in Fig. 6a, PAX molecules were more adsorbed at neutral pH. It can also be seen that at more than 2 nm distances, the number of PAX molecules is less at neutral pH. In general, this diagram shows that the PAX molecules were more around the nanocarrier molecules at neutral pH than at the acidic pH.
Fig. 6
Analyzing the RDF (g(r)) and the gyration radius (a) Density of PAX around the nanocarrier at pH = 5 and 7; (b) Gyration radious change of PAX over time at pH = 5 and 7
Figure 6b also shows the gyration radius, which indicates the radius of the accumulated molecules at various simulation times. According to this diagram, PAX molecules and nanocarriers have accumulated more at neutral pH. This diagram shows that the mean gyration radius and its endpoint at neutral pH were less than at acidic pH. The simulation outputs indicate that PAX molecules are more adsorbed at neutral pH.
Figure 7 demonstrates the information based on the analysis of solvent accessible surface area (SASA). Figure 7a shows the available water area in PAX molecules. PAX molecules were more in contact with water at an acidic pH. Also, the continuous decrease of this diagram at neutral pH shows that PAX molecules at neutral pH were constantly adsorbed to the nanocarrier, while in the acidic state, several fluctuations are seen in the diagram; these fluctuations can be due to the formation of electrostatic repulsion in the acidic state.
Fig. 7
SASA analysis results. a Contact area changes among PAX and water when interacting with the nanocarrier; and (b) Contact surface changes among nanocarrier particles at pH = 5 and pH = 7
Figure 7b also shows the changes in the contact surface between nanocarriers and PAX molecules over time. These changes were compared to the initial simulation mode, which is why the value of this parameter was 0 in the 0-ns mode. The contact area indicates that the surface in contact among the drug molecules and the nanocarrier in the neutral state is nearly twice that of the contact surface among the nanocarrier molecules and the drug in the acidic state. As a result, PAX molecules adsorb in the neutral state was much higher.
Figure 8 also shows the root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) diagrams. These diagrams illustrate the oscillations of the system (separatly for each atom) in terms of time. At the beginning of the simulation, fluctuations are high, while they decrease at the end of the simulation. This trend indicates that the system is moving towards stability. That is, the energy level of the molecules decreases during simulation time. Table 2 also shows the mean, maximum and minimum values of RMSD and RMSF.
Fig. 8
The RMSF and RMSD analysis results. a Fluctuations of PAX atoms upon interacting with the nanocarrier at pH = 5 and pH = 7; (b) fluctuations of PAX particles over time at pH = 5 and pH = 7
Table 2 The mean, maximum and minimum RMSD and RMSF values for PAXFigure 8b and Table 2 show that the mean RMSD at neutral pH was lower than the acidic state, and even the range of fluctuations among maximum and minimum RMSD at neutral pH was smaller than acidic pH. The RMSF analysis also reports the same results for each atom in Fig. 8b and Table 2. According to this analysis, the average fluctuations for all atoms at all time intervals in the neutral pH state were less than in the acidic state. Moreover, Table 2 states that the maximum and minimum fluctuations of RMSF at neutral pH were less than at acidic pH. In addition to showing that the system and the simulation are moving towards a steady state, these analyses also show that this level of stability is better in the neutral state than in the acidic state, and the system is more stable in the neutral state.
The SASA analysis for the Edragit monomer is reported in Fig. 9a. In this analysis, the contact surface of various parts of the monomer with water is shown in color. The range of this contact surface is between 1.03 and 2.34 nm. In the acidic state, the contact surface tends to be more towards 2.34 nm. This means that in the acidic state, the molecules were more inclined to water, and they were able to put more of their surface in contact with water. According to the Arrhenius theory [47], when free hydrogen is released in the acidic state, a charge difference is created between the molecule and water, and they become electrostatically inclined to absorb each other. However, in the neutral state, where the adsorption of water molecules is almost negligible, these monomers are in contact with other molecules. This means that these molecules may have been in contact with PAX, and more adsorption may have occurred.
Fig. 9
SASA analysis results of Edragit and Chitosan monomers. a Changes in contact surface of Eudragit and water monomer atoms at pH = 5 and pH = 7 (b) Contact surface changes of Chitosan and water monomer atoms at pH = 5 and pH = 7
Figure 9b shows the same analyses for the Chitosan monomer. A greater tendency to absorb water could be seen in the acidic state. There is a strong tendency for water to be absorbed by the aromatic ring within the Chitosan monomer in both acidic and alkaline states. This tendency has reached its maximum in the acidic state (2.34 nm). In this case, less adsorption in the acidic state and more in the neutral state could be concluded.
CG-MD simulationIn addition to the AA-MD simulation method, CG-MD simulations have also been performed. These simulations were performed on larger scales and with more molecules than in the AA-MD simulation state. Figure 10 shows the energy diagrams obtained from these analyses. The results of these graphs are pretty similar to the results obtained in the AA-MD method. According to these results, adsorption occurs at neutral and acidic pH, and electrostatic energy causes repulsion among molecules.
Fig. 10
Graph of energy resulting from interactions among PAX and nanocarrier over time at (a) pH = 5, and (b) pH = 7 through CG-MD simulation method
Figure 11 also shows gyration radius and SASA diagrams. The gyration radius in these diagrams is in higher range than in the AA-MD method, while fully confirming the results of that analysis. The result of analysis of the gyration radius indicates that accumulation occurred in the neutral state. The results are also evident in Fig. 11b, which is related to the SASA diagram. The result of SASA analysis in the CG-MD simulation method also dictates PAX adsorption in the neutral state. Based on these results, at neutral pH, the PAX molecules had a lower contact surface with water, meaning that these molecules were attached to the nanocarrier and did not float in water. The simulation snapshots can be seen in Fig. 11 since the scale of the molecules in the CG-MD mode is partially out of the nano mode, and the molecules interact in the micro mode, and their large number and dispersion are visible.
Fig. 11
Results of gyration radius and SASA analysis through CG-MD simulation (a) Changes in the PAX drug gyration radius over time at pH = 5 and pH = 7; (b) PAX and water contact surface changes upon interacting with nanocarrier at pH = 5 and pH = 7; (c) Contact surface changes among nanocarrier particles at pH = 5 and pH = 7
Figure 11c also shows the contact surface among PAX and nanocarriers. According to this diagram, the contact surface among drugs and nanocarriers increases at neutral pH. This contact surface is constant at acidic pH and does not change much during the simulation. Also, repulsions are side by side during simulation.
留言 (0)