
記住我


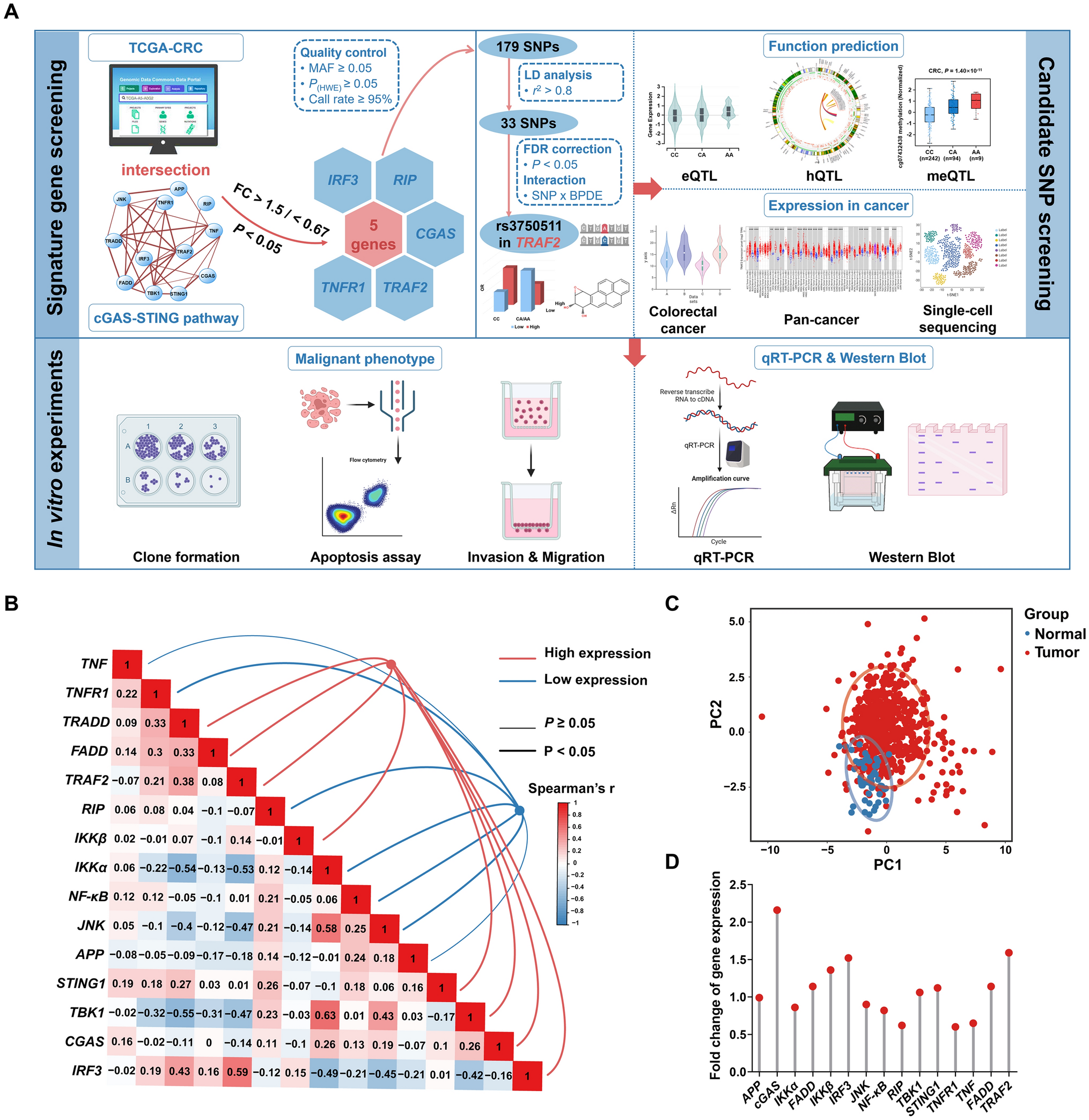


Samples of 24-h urine and blood were collected from 88 healthy adult study participants enrolled between 2017 and 2021 in two studies (Fig. 1). In the RBVD study, 36 vegans and 36 omnivores (sex- and age-matched, 18 females and 18 males in each group, 30 to 60 years old) were recruited in 2017 to compare the influence of the diet on nutrition and biomarker status. Omnivores were included, if they reported to consume at least three servings of meat or two servings of meat and two servings of sausages per week. Vegans did not eat any animal food. In both groups, the dietary habits had to have remained constant for at least 1 year (Weikert et al. 2020). From the 72 study participants examined in 2017, 24 vegans and 26 omnivores followed the invitation for a follow-up examination in 2021. For the assessment of the exposure time trend, only study participants that did not smoke in 2017 and in 2021 were considered (19 omnivores, 20 vegans) (Fig. 1).
Fig. 1
Blood and 24-h urine samples were from 88 adults examined in 2 different studies: The Risks and Benefits of a Vegan Diet (RBVD) study included 72 vegans and omnivores (Weikert et al. 2020), 50 of which were re-examined in 2021, and the Raw Food Eater Study included 16 participants (Abraham et al. 2022)
The goal of the second cross-sectional study was the investigation of internal exposure to heat-induced food contaminants in people strictly abstaining from consumption of food heated to temperatures higher than 42 °C (raw food eater study) (Abraham et al. 2022). The study included 16 healthy subjects (11 males and 5 females, age between 20 and 65 years) following a raw food diet for at least four months (Fig. 1). Exclusion criteria were any consumption of beverages like coffee or tea or of hot meals and smoking.
The participants collected their urine completely for 24 h using preservative-free plastic containers provided during the first study visit, starting the day before the second study visit and ending with the first urine in the morning of the second visit. The samples were thoroughly mixed, weighed, aliquoted and stored at − 80 °C until analysis. Blood samples were taken using EDTA tubes S-Monovette® (9 mL, Sarstedt, Numbrecht, Germany), centrifuged (2,500⋅g, 12 min) and the plasma was removed. The erythrocytes were washed twice with 0.9% aqueous sodium chloride (2.5 mL) and resuspended with 2.5 mL of water. The Hb content was determined with a HemoCue Hb 201 + analyzer (Radiometer, Willich, Germany) and the erythrocyte samples were stored at − 80 °C.
The RBVD study (no. EA4/121/16 and EA4/063/21) and the raw food eater study (No. EA4/040/19) were approved by the Ethics Committee of Charité University Medical Center Berlin. The raw food eater study (No. 00017436) and the RBVD follow-up study (No. 000 25857) were registered in the German Clinical Trials Register (DRKS). The studies were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments (Abraham et al. 2022; Weikert et al. 2020).
All participants got detailed oral consultations about the rationales and the methods of the studies and gave informed consent in writing after their first visit of the study center.
ChemicalsPotassium hydrogen carbonate, aqueous ammonia (25%), phenylboronic acid (> 97%), sodium sulfate (anhydrous, granulated, for organic trace analysis), acetic acid, hydrochloric acid (13 N), diethyl ether, ethyl acetate, tert-butyl methyl ether (tBME), iso-hexane, iso-octane, methanol, and acetonitrile (for UHPLC-MS) were purchased from Merck KGaA (Darmstadt, Germany). Formic acid (≥ 96%), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and fluorescein-5-isothiocyanate (FITC, isomer I, > 95%) were provided by Sigma (Steinheim, Germany), and water (UHPLC MS-Optigrade) was supplied by LGC Standards GmbH (Wesel, Germany). Sodium sulfate was dried overnight in a muffle furnace (~ 200 °C) before use. All reagents and solvents were of analytical grade.
The isotope-labeled standard 3-chloro-1,2-propanediol-d5 (3-MCPD-d5) was obtained from Sigma-Aldrich (Steinheim, Germany) and 2-chloro-1,3-propanediol-d5 (2-MCPD-d5) was from LGC Standards GmbH. The quantification standard 3-(fluorescein-5-yl)-1-(2,3-dihydroxypropyl)-5-d7-isopropyl-2-thioxo-4-imidazolidinone (DHP-d7-Val-FTH) and the dipeptide N-(2,3-dihydroxypropyl)-Val-Leu-anilide (DHP-VL-An) were synthesized as described by Hielscher et al. (2017) and Abraham et al. (2019), respectively.
Preparation of standard solutionsAmounts of ~ 1 mg of DHP-d7-Val-FTH or of the dipeptide DHP-VL-An were weighed and dissolved in respective volumes of DMSO to yield solutions of 5 mmol/L. The working solutions of DHP-d7-Val-FTH (50 nmol/L) or of the dipeptide DHP-VL-An (100 nmol/L) were prepared by dilution in water/acetonitrile (1:1), aliquoted for further use and stored at − 80 °C.
Two separate stock solutions of 2-MCPD-d5 and 3-MCPD-d5 (1 g/L each) were prepared by dissolution of 10 mg 2-MCPD-d5 or 10 mg 3-MCPD-d5 in 10 mL methanol. The working solution (1 µg/L each of 2-MCPD-d5 and 3-MCPD-d5) was prepared by mixing 1 mL each of the stock solutions with 998 mL methanol. The working solution was aliquoted into 10 mL screw cap vials which were sealed with laboratory film and stored at room temperature.
Edman degradation and solid-phase extraction (SPE) of DHP-Val-FTHDHP-Val was cleaved from the N-termini of Hb using a modified Edman degradation with FITC as described (Fig. S1) (Gauch et al. 2022; Rydberg et al. 2009). Aliquots of erythrocyte samples containing 35 mg Hb were mixed with 15 µL of 1 M aqueous potassium hydrogen carbonate, 10 µL of the isotope-labeled standard solution containing DHP-d7-Val-FTH (50 nmol/L), and 5 mg FITC in 30 μL DMF. After incubating for 18 h at 37 °C, protein was precipitated by addition of 1.6 mL acetonitrile and the samples were centrifuged (18,000⋅g, 10 min). The supernatant was pH-adjusted with 25 µL of 1 M aqueous ammonium hydroxide and transferred to an Oasis MAX cartridge (60 mg; Waters, Eschborn, Germany), preconditioned with 2 mL acetonitrile and 2 mL water. The cartridge was washed with 2 mL each of acetonitrile, water and 1% aqueous formic acid, and DHP-Val-FTH was eluted with 3 mL acetonitrile/water (9:1) containing 1% formic acid. The eluate was evaporated to dryness and reconstituted in 50 µL acetonitrile/water (1:1) containing 1% formic acid.
UPLC–MS/MS analytical quantification of DHP-Val-FTHThe UPLC-MS system comprised an Acquity I-Class (Waters) connected to a triple quadrupole-hybrid ion trap mass spectrometer QTrap6500 (Sciex, Darmstadt, Germany) equipped with an electrospray ionization source operated in the positive mode. The DHP-Val-FTH (10 µL sample volume) was separated chromatographically using an Acquity HSS T3 column (1.8 µm, 2.1 mm × 150 mm, Waters). The eluent solvents were water + 0.1% formic acid (eluent A) and acetonitrile + 0.1% formic acid (eluent B). A two-step gradient was applied at a flow rate of 0.4 mL/min: 0–1 min (10% eluent B), 1–15 min (10–50% eluent B), 15–21 min (50–70% eluent B), 21–22.5 min (90% eluent B) and 22.5–24 min (10% eluent B). DHP-Val-FTH and DHP-d7-Val-FTH were recorded by multiple reaction monitoring (MRM). Table S1 in the Supplemental Information summarizes the fragmentation transitions and the respective parameters (declustering potential, entrance potential, collision energies and cell exit potentials). The other mass spectrometric parameters were set to the following values: curtain gas, 20 psi; ion source temperature, 450 °C; ion spray voltage, 5500 V; ion source gas 1, 60 psi; ion source gas 2, 50 psi; collision-activated dissociation gas, medium. The data were recorded and analyzed with Analyst 1.7.1 Software (Sciex).
The adduct levels were calculated using Eq. 1:
$$_\left[\frac\right]=\frac_}_} * _ \left[pmol\right] }_ \left[g\right]}$$
(1)
with Aanalyte and AIS as the peak areas of the quantifier signals of the analyte and of the internal standard, respectively, and with amountIS and amountHb as the quantities of the internal standard and of Hb applied in the Edman degradation, respectively. The accuracy of quantification of DHP-Val was improved using ten control samples of an erythrocyte pool (incorporated in each sample set). Five of these samples were spiked with 10 µL of 100 nmol/L DHP-VL-An and otherwise worked up as described. These samples allowed determining the efficiency of the FITC-mediated Edman degradation, which was used to correct the DHP-Val levels determined in the human samples (Abraham et al. 2019; Monien et al. 2020). The limits of detection (LOD) and quantification (LOQ) were 0.25 and 0.71 pmol/g Hb. Further details on the validation for the determination of DHP-Val in Hb described previously (Gauch et al. 2022; Rydberg et al. 2009) are summarized in Table S2 of the Supplemental Information.
Extraction and derivatization of 2/3-MCPDExtraction from urine samples and derivatization of 2/3-MCPD using phenylboronic acid were described before (Abraham et al. 2021). Briefly, urine samples were thawed, mixed vigorously, and aliquots of 4 mL were spiked with 40 μL each of 3-MCPD-d5 (1 µg/L) and of 2-MCPD-d5 (1 µg/L). The samples were washed with 3.5 mL of tBME/isohexane (1:4). 2/3-MCPD were extracted using 2.5 mL of diethyl ether/ethyl acetate (9:1). Phase separation was completed by a 2-min centrifugation (3,500 rpm), and the extraction was repeated twice. The combined organic layers were dried over sodium sulfate, mixed with 50 μL of phenylboronic acid (2 mg/mL) in diethyl ether/ethyl acetate (9:1), and concentrated in a moderate nitrogen flow. A residual volume of 1 mL was dried with 200 mg sodium sulfate and the solutions were concentrated to dryness. The residuals were taken up in 200 μL of iso-octane, transferred into an analysis vial with a 200 µL-insert and stored in the freezer prior to GC–MS analysis. Analyses were carried out in duplicate.
GC–MS analysis of 2/3-MCPDThe dioxaborolane derivatives of 2/3-MCPD (Fig. S2) were analyzed by an accredited laboratory (SGS Germany GmbH, Hamburg, Germany) using a gas chromatograph 7890B system (Agilent Technologies, Santa Clara, CA, USA) equipped with a Programmable Temperature Vaporizor (PTV) injection device. The following conditions were applied: injection volume 2–4 μL (pulsed splitless injection), PTV temperature program: 80 °C (isothermal for 0.1 min), increase with 2 °C/s to 140 °C (isothermal for 6 min), increase with 10 °C/s to 320 °C (isothermal for 10 min). PTV purge gas flow: 50 mL helium/min at 0.5 min to 1 min (septum purge 3 mL helium/min). Samples were separated on a Rxi-17 (Resteck GmbH, Bad Homburg, Germany) with a stationary phase of 50% diphenyl/50% dimethylpolysiloxane (30 m × 0.25 mm, 0.25 mm film thickness) and the pre-column HP-5 ms (Agilent Technologies) with a stationary phase of 5% phenyl/95% dimethylpolysiloxane (2.4 m × 0.32 mm, 0.25 mm film thickness). Helium was used as carrier gas at a constant flow rate of 1.4 mL/min. The oven temperature started at 80 °C (isothermal for 2 min) followed by a gradient of 5.4 °C/min up to 150 °C (isothermal for 4 min). Then, the temperature increased with 20 °C/min to 280 °C (isothermal for 5 min). Analytes were ionized by electron impact and detected using the single quadrupole mass spectrometer 5977B (Agilent Technologies) operated in the selected ion monitoring (SIM) mode. Mass-to-charge ratios (m/z) of the detected phenylboronic derivatives of the analytes were m/z = 146, 147, 196, 198 (3-MCPD), m/z = 149, 150, 201, 203 (3-MCPD-d5), m/z = 196, 198 (2-MCPD). 3-MCPD was quantified from the respective signal of the 3-MCPD-d5 phenyl boronic acid derivative using the ratio of the traces 147/150. 3-MCPD-d5 (m/z = 150) was also applied for the quantification of 2-MCPD (m/z = 196) with the help of a specific correction factor (2.47). Every eighth sample was a quality control sample (human urine with defined amounts of 2/3-MPCD). Ratios of the response factors, e.g., m/z = 150 vs. 147 (3-MCPD-d5 vs. 3-MCPD), were checked for constancy. The LOD and LOQ values were 0.1 and 0.25 μg/L for 3-MCPD, respectively, and 0.12 and 0.3 μg/L for 2-MCPD, respectively. Further validation data on 2/3-MCPD analyses in urine samples described by Abraham et al. (2021) are summarized in Table S3 of the Supplemental Information. The analytesʼ molecular structures are depicted in Supporting Information Fig. S1. Analyses were performed in duplicates.
Reverse dosimetry estimations of daily intakes of 2/3-MCPD and glycidolThe daily oral intake of 2/3-MCPD (free or in form of fatty acid esters) was estimated from the excretion of 2/3-MCPD in 24-h urine samples using the ratio between the excreted amounts and the oral exposure (2-MCPD: 14.3 ± 3.1%; 3-MCPD: 3.73 ± 0.95%, mean ± SD) determined in 12 adult participants after the controlled exposure to a hazelnut oil with known high amounts of 2/3-MCPD fatty acid esters (Abraham et al. 2021). The glycidol intake was estimated from the DHP-Val levels. Assuming steady-state conditions for adduct formation after at least four months of continuous glycidol exposure, the daily exposure (D, µg/kg bw/day) can be calculated by Eq. 2,
$$D \left[\frac\right]=\frac\right] }/\frac\right]}$$
(2)
with the adduct level (H), and the constants (τ = 104 d, mean lifetime of the adducts) and the mean adduct level/dose ratio (k = 0.082 ± 0.004 pmol DHP-Val/g Hb per μg glycidol/kg bw) determined in 11 adult participants after controlled exposure to glycidol fatty acid esters for 28 days (Abraham et al. 2019).
Statistical analysisDue to the influence of tobacco smoking on the exposure of 2/3-MCPD as well as glycidol, the data of smoking and non-smoking participants were evaluated separately. Daily urinary excretion of 2/3-MCPD and DHP-Val in Hb are reported as median values with the inter-quartile range (IQR) in brackets. Differences of biomarker levels were evaluated with the Mann–Whitney rank-sum test, and differences between biomarker levels determined in 2017 and 2021 were evaluated with a Wilcoxon signed rank test using SigmaPlot 14.0 (Systat Software, Inc., Erkrath, Germany). Differences with p-values < 0.05 were considered statistically significant. Spearman rank order correlations were calculated with SigmaPlot 14.0 (Systat Software, Inc.).
留言 (0)