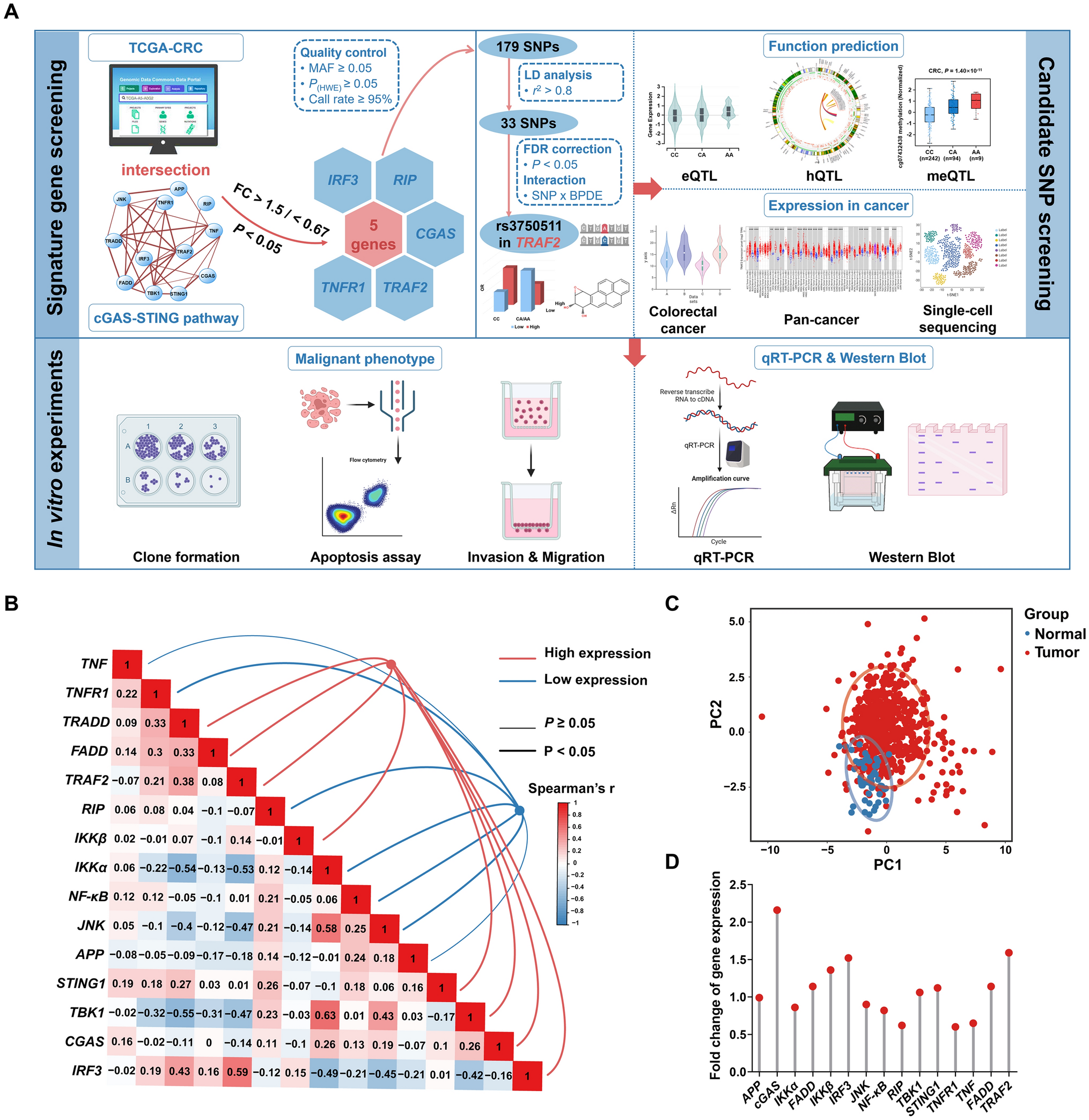
Materials
Cell culture media and supplements used for culturing Ishikawa cells and THP1-Lucia™ NF-κB monocytes were purchased from Gibco® Life Technologies (Karlsruhe, Germany), except for charcoal–dextran stripped (CD-) FBS which was acquired from Fisher Scientific (Catalog #A3382101). 17β-Estradiol (E2) and 4-nitrophenyl phosphate were purchased from Sigma-Aldrich (Schnelldorf, Germany), while the lipopolysaccharide (LPS; from E. coli) and dexamethasone (Dexa) were acquired from Sigma-Aldrich (Steinheim, Germany). Normocin, zeocin and Quanti-Luc™ were from Invivogen (Toulouse, France). Triton®X 100 and dimethyl sulfoxide (DMSO) were purchased from Carl Roth GmbH&Co (Karlsruhe, Germany). Reference standards of Alternaria toxins were obtained from several suppliers or kindly provided by other researchers. Interested readers may refer to Puntscher et al. (2019a) for details. The mycotoxins AOH, ATX-I, ALTP and TeA, tested for their immunomodulatory and/or antiestrogenic effects, were acquired from Sigma-Aldrich (Schnelldorf, Germany), Szabo-Scandic (Vienna, Austria), Cfm Oskar Tropitzsch (Marktredwitz, Germany), and Santa Cruz Biotechnology (Heidelberg, Germany), respectively. For LC–MS/MS analyses, acetonitrile (ACN) and water (both LC–MS grade) were purchased from Honeywell (Seelze, Germany). For the SFC-based fractionation of CE, compressed CO2 (4.5 grade, purity ≥ 99.995%) was purchased from Messer (Frankfurt am Main, Germany) and methanol (ultrahigh-gradient grade) was purchased from Merck (Darmstadt, Germany).
Complex extract of Alternaria mycotoxins and AOH-spiked Alternaria extract
The CE employed in the present study was previously obtained by growing the Alternaria alternata strain DSM 62010 on long rice for 21 days, and chemically characterized by LC–MS/MS analysis (Puntscher et al. 2019a, b). Concentrations of the mycotoxins in CE at the highest concentration applied in the NF-κB assay (20 µg/mL) are reported in Table 1. To assess the contribution of AOH to the immunosuppressive properties of CE (characterized by a low concentration of the mycotoxin), an AOH-spiked Alternaria extract (SE) was prepared by dissolving pure AOH in CE, such that 20 µg/mL of CE contained 20 µM AOH, 10 µg/mL of CE contained 10 µM AOH, and so forth. Prior to the experiments, the SE was analyzed by LC–MS/MS to ensure the correct addition of AOH to the extract.
Table 1 Concentration of Alternaria mycotoxins to which THP1-Lucia™ cells were exposed during treatment with the highest CE concentration tested (20 µg/mL)Fractionation of the Alternaria extract by supercritical fluid chromatography
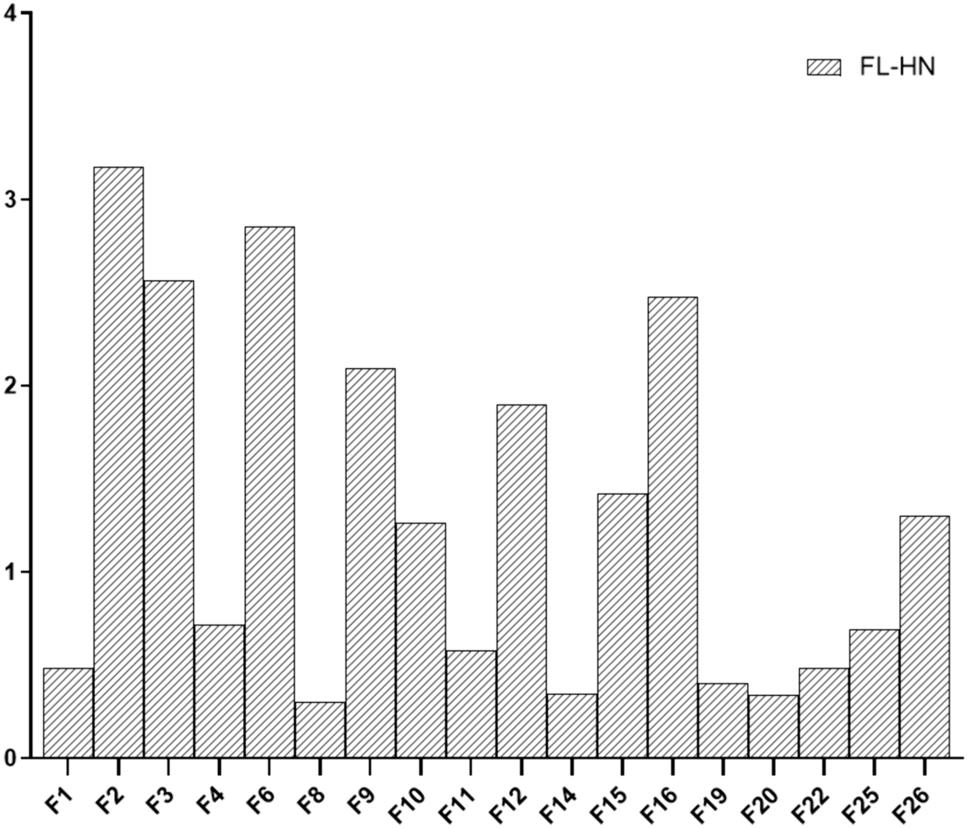
For the fractionation of the CE, a semi-preparative SFC was used (Prep-15 SFC system, Waters, Milford, MA, USA). The system comprises a fluid delivery module connected to an Accel 500 LC chiller, a Waters 2767 sample manager, a ten-port column oven, a back pressure regulator, a heat exchanger, a make-up pump, a Waters 2998 PDA, and a Waters 2424 ELSD. The system was operated by the software MassLynx V4.1 and FractionLynx. A Torus 1-Aminoanthracene column (5 μm, 10 mm × 250 mm; Waters Milford, MA, USA) was used at a column temperature of 40 °C with pure CO2 as solvent A and methanol as solvent B applying the following gradient: 0–4.0 min from 5 to 50% B; 4.0–5.0 min 50% B; 5.0–6.0 min ramp down to 5% B, and 6.0–7.0 min 5% B as equilibration step. For fractionation CE was diluted with a solvent composed of 70% hexane and 30% isopropanol (v/v) prior to injection (100 µL). The flow rate was 15 mL/min, the backpressure was set to 120 bar and after the column the eluents were combined with a methanol make-up flow of 5 mL/min to facilitate the detection and fraction collection. The retention time stability was checked each time prior fraction collection. A representative chromatogram, recorded at a wavelength of 205 nm, indicating the sampling events is presented in Supplementary Figure S1. Separation performance was monitored with an ultra-high-performance supercritical fluid chromatograph (UHPSFC, Acquity UPC2-DAD/ELSD/MS, Waters, Milford, MA, USA). The eleven collected fractions (F1-F11) were dried under nitrogen flow at room temperature, followed by resuspension in DMSO to obtain stock solutions with a concentration of 3 mg/mL.
LC–MS/MS analysis for the quantification of mycotoxins contained in the CE-fractions
For the quantification of mycotoxins in the eleven collected CE-fractions, LC–MS/MS analyses were performed using the method developed by Puntscher et al (2018). This method has been validated for the analysis of mycotoxins in food matrices and further applied for the analysis of urine, feces (Puntscher et al. 2019a, b), and cell culture media (Crudo et al. 2020). Briefly, CE-fractions were diluted with a solvent consisting of 50% acetonitrile and 50% water (v/v) and analyzed by using a high-performance liquid chromatographic system (HPLC, UltiMate3000, Dionex Thermo Fisher Scientific, Vienna, Austria) coupled to a TSQ Vantage triple quadrupole mass spectrometer equipped with a heated electrospray ionization (HESI) interface (Thermo Fisher Scientific). For the chromatographic separation, a Supelco Ascentis Express column (C18, 2.7 μm, 10 cm × 2.1 mm) equipped with a Phenomenex SecurityGuard™ precolumn (C18, 2 mm, Phenomenex, Torrance, CA) was used. Data were acquired in negative electrospray ionization mode. Further details are reported in Puntscher et al. (2018).
To avoid carry-over phenomena and verify the overall performance of the instrument, solvent blanks were routinely injected. Samples were randomly analyzed, and quantification of the analytes was performed by external calibration (calibration set injected after every approximately 20 samples). For instrument control, data acquisition and data evaluation, Chromeleon™ chromatography data system software (v. 6.80 SR13 Build 3818), Xcalibur™ software (v. 3.0, Thermo Scientific), and TraceFinder™ software (v. 3.3, Thermo Scientific) were used, respectively.
NF-κB gene reporter assay
THP1-Lucia™ NF-κB monocytes, obtained by transfection of THP-1 cells with an NF-κB–inducible luciferase reporter construct, were purchased from InvivoGen (Toulouse, France) and cultivated following the provider’s instructions. Briefly, cells were routinely maintained in Roswell Park Memorial Institute (RPMI)-1640 medium containing 25 mM HEPES buffer, 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1% penicillin/streptomycin solution (100 U/mL) and 100 µg/mL normocin. Cells were sub-cultivated twice a week and, to maintain selection pressure, 100 µg/mL zeocin were added to the flask every other passage.
For the experiments, 1 × 105 cells/well were seeded into 96-well plates and simultaneously exposed to the extracts (CE and SE; 0.0001–20 µg/mL), fractions of CE (F1–F11; 0.075–7.5 µg/mL), single mycotoxins (TeA, 0.1–250 µM; AOH, 0.0001–20 µM; ATXI and ALTP, 0.001–20 µM), solvent control (0.25% DMSO), or negative control (1 µM Dexa). For the positive and negative control treatments, 10 ng/mL LPS from E. coli were added to the cells 2 h after the start of the incubation. To assess the ability of the test compounds/extracts/fractions to activate the NF-κB pathway, cells were incubated for 20 h at 37 °C and 5% CO2 without the addition of LPS. On the contrary, their immunosuppressive properties were tested by adding 10 ng/mL LPS (after 2 h, as for the positive control) and incubating the cells for a further 18 h. The DMSO concentration in the various treatments was always 0.25%. At the end of the incubation time (20 h), the luciferase activity was assessed by employing the coelenterazine-based luminescence reagent “Quanti-Luc™”. Briefly, the plate was centrifuged at 140 rcf for 2 min, after which 10 µL of the cell supernatants were transferred to a white 96-well plate. The luminescence measurement was performed, after the automatic addition of 50 µL/well of the detection reagent, with a microplate reader equipped with an injection system (BioTec Synergy H1; Agilent Technologies, Santa Clara, USA). Every condition was tested in three–six technical replicates and in at least three independent experiments.
Alkaline phosphatase assay
To assess the estrogenic and antiestrogenic properties of the CE-fractions (0.015–1.5 µg/mL) and single mycotoxins (ATX-I and ALTP; 0.0002–10 µM), the human endometrial adenocarcinoma Ishikawa cell line was employed (European Collection of Authenticated Cell Cultures; Wiltshire, United Kingdom). This estrogen-sensitive cell line was chosen as a model system because of the constitutive expression of both ERα and ERβ (Boehme et al. 2009). Cells were routinely maintained at 37 °C and 5% CO2 in Minimum Essential Medium containing phenol red and supplemented with 5% FBS (v/v), 1% penicillin/streptomycin (v/v; 100 U/mL) and 1% L-glutamine (v/v). Cells were sub-cultured every third to fourth day at a confluency of approximately 80%.
For the AlP assay, cells in assay medium (DMEM/F-12 without phenol red supplemented with 5% charcoal/dextran–treated FBS) were seeded in 96-well plates at a density of 1 × 104 cells/well, followed by incubation at 37 °C and 5% CO2 for 48 h prior to cell treatment. As positive and solvent controls, 1 nM E2 and 0.1% DMSO were used, respectively. To assess the antiestrogenic properties of the various test conditions, cells were co-incubated with 1 nM E2, while no E2 was added for the assessment of the estrogenic properties. All conditions were tested in at least three technical replicates and in three independent experiments. After 48 h exposure of cells to the various test conditions (with and without co-incubation with 1 nM E2), the cells were washed three times with phosphate-buffered saline (PBS) and lysed by shock-freezing at −80 °C for 20 min. Cells were thawed at room temperature for 5 min, followed by the addition of 50 µL/well of an AlP buffer containing 5 mM 4-nitrophenylphosphate, 1 M diethanolamine, and 0.24 mM MgCl2 (pH 9.8). After 5 min of incubation at room temperature, the AlP activity was determined using a microplate reader (BioTec Synergy H1; Agilent Technologies, Santa Clara, USA) by measuring the absorbance at 405 nm every 2 min for 1 h. The slope of the curves within the linear range was calculated as a measure of the enzyme’s activity.
Celltiter-blue® (CTB) cell viability assay
To assess the impact of test compounds/extracts/fractions on the viability of THP1-Lucia™ and Ishikawa cells, the CTB cell viability assay was performed. For THP1-Lucia™ cells, two hours before the end of the incubation time, a positive control for cytotoxicity was prepared by exposing untreated cells to 0.01% Triton X. At the end of the incubation period, the CTB reagent was added to each well of the plate at a 1:10 dilution, and the cells were then incubated at 37 °C and 5% CO2 for an additional 2 h. Plates were centrifuged at 140 rcf for 2 min, followed by transferring 100 µL of cell supernatants to a black 96-well plate. Ishikawa cells were exposed to the various test conditions and controls for 48 h. Afterwards test media were replaced with 100 µL/well of CTB solution (CTB reagent diluted 1:10 with phenol red-free DMEM/F12 medium). As a positive control served 0.005% Triton-X ®. After 50 min incubation at 37 °C and 5% CO2, 100 µL of cell supernatants were transferred to a black 96-well plate.
For both cell lines, the fluorescence intensity was measured at an excitation wavelength of 530 nm and an emission wavelength of 560 nm using a microplate reader. Every condition in each experiment was tested in three technical replicates, and the data were reported as the mean value (+ standard deviation) of at least three independent experiments.
Statistical analysis
Independent Student t-test was performed to determine significant differences between the treatments and the respective positive or solvent controls. To assess significant differences among the various concentrations of each tested mycotoxin, extract, or fraction, analysis of variance (one-way ANOVA) with Fisher-LSD post hoc test was performed. Samples were considered significantly different for *p < 0.05, **p < 0.01, or ***p < 0.001. All statistical tests were conducted using OriginPro 2021 software (v. 9.8.0.200, OriginLab Corporation, Northampton, MA, USA).





留言 (0)