
記住我




Prior studies have established the role of Kindlin-2 as a major driver of tumor progression and metastasis in several cancers including the one that originates in the breast [14]. Published studies form our group and others have also shown that Kindlin-2 is involved in the activation of the oncogenic behavior of TNBC tumors, both in vitro and in vivo [15, 16, 19, 21,22,23,24, 26, 27, 29]. Interrogation of a BC tumor microarray generated from BC specimens representing the different BC subtypes [15, 30], showed high levels of Kindlin-2 staining in advanced BC stages (Fig. S1A, B). Kindlin-2 staining score was significantly (p < 0.05) higher in BC tumors compared to normal breast tissues (Fig. S1B). More importantly, Kindlin-2 staining score was significantly (p < 0.05) higher in hormone receptor-negative and Her2-negative tumors, e.g. TNBC subtype (Fig. S1B), a tread that was also found in human and murine cell lines of TNBC nature [15, 30]. These findings were further confirmed by interrogation of public BC datasets from Oncomine (www.oncomine.org), where we found Kindlin-2 mRNA expression levels to be significantly (p < 0.001) higher in BC tumors compared to normal breast tissues [15]. Furthermore, interrogation of the KM-Plotter BC database (https://kmplot.com/analysis/) showed increased mRNA expression levels of Kindlin-2 correlate with poor disease outcome in patients with BC tumors, irrespective of subtype (Fig. S1C and [15]), and specifically in patients with TNBC tumors, at the mRNA levels (Fig. S1D), and at the protein levels (Fig. S1E). These findings, together with our published studies [15, 16, 21,22,23, 29], support the key role that Kindlin-2 plays in the pathology of BC tumors, in general, and in TNBC tumors, in particular.
One of the major functions of Kindlin-2 is the activation of the inside-out signaling of integrins through its physical interaction with the cytoplasmic tail of several integrin β-subunits, including β1-Integrin (Reviewed in [25, 31]). Additionally, we previously showed that Kindlin-2 activates the CSF1/EGF paracrine oncogenic loop in TNBC through the regulation of TGF-β signaling [15]. Interestingly, a study by Wei et al. [28] showed that Kindlin-2 also interacts with the cytoplasmic region of TGF-β type one receptor (TβRI). Accordingly, our published studies have shown that Kindlin-2 plays a major role in the regulation of TNBC tumor progression and metastasis through the regulation of the oncogenic activities of both Integrins and TGF-β. Based on this information we sought to investigate the molecular mechanisms that regulate the Kindlin-2 interaction with both β1-Integrin and TβRI, and the role of these interactions in the regulation TNBC tumor progression and metastasis.
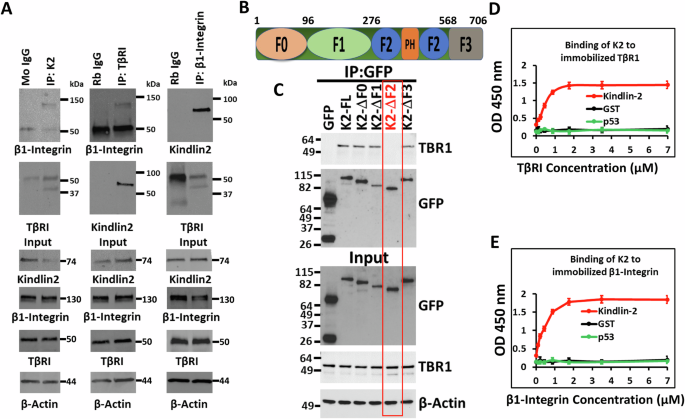
Kindlin-2 interacts with both β1-Integrin and TβRICo-immunoprecipitation using total protein lysates from MDA-MB-231 TNBC cells showed Kindlin-2 immunocomplexes captured both TβRI and β1-Integrin (Fig. 1A, left panels), while TβRI immunocomplexes also readily captured both Kindlin-2 and β1-Integrin (Fig. 1A, middle panels), and β1-Integrin immunocomplexes readily captured Kindlin-2 and TβRI (Fig. 1A, right panels), thereby implicating Kindlin-2 as a potential adapter that coordinates the formation of TβRI and β1-Integrin complexes. In addition, pulldown assays identified the F2 domain within Kindlin-2 (Fig. 1B) as being necessary for the interaction between Kindlin-2 and TβRI (Fig. 1C). The interaction between Kindlin-2 and the cytoplasmic tail of β1-Integrin has already been established to be mediated via the Q614W615 amino acid doublet that resides within the F3 domain of Kindlin-2 [32]. Finally, solid binding assay monitoring the binding of recombinant Kindlin-2 to immobilized TβRI (Fig. 1D) or β1-Integrin (Fig. 1E), established a direct binding of Kindlin-2 to TβRI and β1-Integrin, thereby establishing a physical bridge between TβRI and β1-Integrin.
Fig. 1: Kindlin-2 interacts with both β1-Integrin and TβRI.
A Co-immunoprecipitation of total protein lysates from MDA-MB-MB-231 cells showing interactions between endogenous K2, β1-Integrin and TβRI proteins. B Diagram representation of the different K2 domains. C Pull-down assays showing that the F2 domain of K2 is necessary for the interaction between K2 and TβRI. Solid binding assay monitoring the binding of recombinant Kindlin-2 to immobilized TβRI (D) or β1-Integrin (E).
Kindlin-2 is required for the stabilization of the β1-Integrin:Kindlin-2:TβRI protein complexProbing further into the importance of Kindlin-2 in maintaining the integrity of β1-Integrin/Kindlin-2/TβRI protein complexes, we found loss of expression of Kindlin-2 (K2-KO) in MDA-MB-231 (Fig. 2A) or 4T1 (Fig. 2B) TNBC cells leads to the degradation of both TβRI and β1-Integrin proteins. mRNA expression levels of either TβRI or β1-Integrin were not significantly affected in the K2-KO MDA-MB-231 (Fig. 2C) or 4T1 (Fig. 2D) TNBC cells, suggesting that loss of expression of TβRI and β1-Integrin in the K2-KO cells was a result of protein degradation, but not at the mRNA transcription levels. We also used flow cytometry to measure the cell surface expression levels of β1-Integetrin and found loss of expression (KO) of Kindlin-2 resulted in inhibition of β1-Integrin expression at the surface of the cell membrane where it exerts its signaling functions (Fig. 2E). Over-expression of full-length Kindlin-2 (K2-full) in the K2-KO MDA-MB-231 cells restored cell surface expression of β1-Integrin to levels comparable to those found in the control cells (Fig. 2E, yellow histogram). We also used the HUTS4 assay that measures the active state of β1-Integrin [33], and found K2-KO, resulted in inhibition of activation levels of β1-Integrin (Fig. 2F), which could also be restored by over-expressing of full-length Kindlin-2 (K2-full) in the K2-KO cells (Fig. 2F, yellow histogram). In both experiments, we used β1-Integrin-KO as a control for both surface expression and activity. Moreover, treating the K2-KO MDA-MB-231 (Fig. 2G) or 4T1 cells (Fig. 2H) with the proteasome inhibitor MG132 resulted in the restoration of both TβRI and β1-Integrin proteins to levels found in the control cells, meanwhile overexpression of full-length Kindlin-2 in the K2-KO cells restored protein expression of both TβRI or β1-Integrin (Fig. 2I, J). Therefore, these data support the role of Kindlin-2 in maintaining the physical integrity of the TβRI:Kindlin-2:β1-Integrin protein complex.
Fig. 2: Kindlin-2 is required for the stabilization of the β1-Integrin:Kindlin-1:TβRI protein complex.
MDA-MB-231 cells (A) and 4T1 cells (B) were subjected to CRISPR/Cas 9 mediated gene editing to knockout FERMT 2 (K2), ITGB1 (β1-Integrin) or TβRI genes, and their protein expression was assessed by Western Blot analysis. β-Actin is a loading control. qt-RT-PCR of mRNA expression levels of TβRI and β1-Integrin in the K2-KO MDA-MB-231 (C) and 4T1 (D) TNBC cells. E, F Flow cytometry histograms of the cell surface expression levels of β1-Integetrin (E) and its activated form (F) in K2-KO, ITGB1-KO and K2-full rescued K2-KO MDA-MB-231 cells. Western Blot analysis with indicated antibodies showing expressions of TβRI and β1-Integrin proteins in MDA-MB-231 K2-KO after treatment with MG132 (G, H) or after rescue of K2-full length (I, J). The numbers under each WB band represent the fold change in signal intensity with respect to its respective control band in each panel after normalization to the loading control signal. Data shown are representative of 3 replicates. Data are the mean ± SD (n = 3, *p < 0.05, Student’s t-test).
Loss of expression of either Kindlin-2, TβRI or β1-Integrin inhibits the in vitro oncogenic behavior of TNBC tumors, and re-expression of Kindlin-2 is sufficient for the restoration of these oncogenic activitiesTo investigate the biological significance of loss of expression of either Kindlin-2, TβRI or β1-Integrin (ITGB1), and their effect on the oncogenic behavior of TNBC cells, we performed several in vitro assays. Parental MDA-MB-231 or 4T1, or their Kindlin-2-, TβRI- or ITGB1-deficient (KO) derivatives were subjected to the wound healing assay (Fig. 3A–D). Loss of expression of either Kindlin-2, TβRI or ITGB1 resulted in the inability of the MDA-MB-231-KO cells (Fig. 3A, B) or the 4T1 KO cells (Fig. 3C, D) to effectively close the scratch wound after 24 h, supporting the role of Kindlin-2, TβRI and β1-Integrin in cancer cell migration. Loss of expression of either Kindlin-2, TβRI or ITGB1 also inhibited the colony formation potential, a hallmark of cancer cells phenotype, of both MDA-MB-231 (Fig. 3E, F) and 4T1 (Fig. 3H) cells. In addition, to mimic the behavior of cancer cells in the tumor microenvironment, we performed 3D-tumorsphere growth and tumorsphere invasion of extracellular matrices (ECMs). 3D-tumorsphere growth was significantly (p < 0.01) inhibited in both the MDA-MB-231 (Fig. 3I, J and Fig. S2A) and the 4T1 (Fig. 3, L and Fig. S2B) cells deficient in either Kindlin-2, β1-Integrin or ITGB1 K2-KO. Similarly, MDA-MB-231 or 4T1 cells (Fig. 3M, N and Fig. 3O, P, respectively) were unable to invade ECM containing Matrigel (Fig. 3M–P, and Fig. S3A, B). Interestingly re-expression of full-length Kindlin-2 (K2-Full) in K2-KO MDA-MB-231 or 4T1 cells restored the cell migration potential of MDA-MB-231 cells (Fig. 4A, B) and 4T1 cells (Fig. 4C, D). Colony formation activity was also restored in the K2-KO MDA-MB-231 and 4T1 cells re-expressing K2-Full (Fig. 4E, F and Fig. 4G, H, respectively). In a similar manner, the ability of MDA-MB-231 and 4T1 cells to establish tumorspheres in 3D organoid growth assays (Fig. 4I–L, and Fig. S4A, B) and for these tumorspheres to invade ECMs (Fig. 4M-P, and Fig. S5A, B) were also restored in the K2-KO MDA-MB-231 and 4T1 cells re-expressing full-length Kindlin-2. These data have so far demonstrated the requirement of Kindlin-2, TβRI and ITGB1 for the common oncogenic activities of cancer cells, and for Kindlin-2 to be sufficient to restore these activities downstream of either β1-Integrin and TβRI.
Fig. 3: Loss of expression of either Kindlin-2, TβRI or β1-Integrin inhibits the in vitro oncogenic behavior of TNBC tumors.
Wound healing assay (A–D), 2D-colony formation assay (E–H), 3D-tumorsphere growth assay (I–L), and 3D-tumorsphere invasion assay (M–P) of control (Ctrl) MDA-MB-231 and 4T1 cells and their K2-KO, TβRI-KO and ITGB1-KO derivatives. Scale bar: 100 µm. Data are the mean ± SD (n = 3, **p < 0.01; ***p < 0.001, Student’s t-test).
Fig. 4: Re-expression of Kindlin-2 expression rescues the in vitro oncogenic behavior of TNBC tumors.
Wound healing assay (A–D), 2D-colony formation assay (E–H), 3D-tumorsphere growth assay (I–L), and 3D-tumorsphere invasion assay (M–P) of control (Ctrl) MDA-MB-231 and 4T1 cells, and their K2-KO or K2-KO rescued with K2-full derivatives. Scale bar: 100 µm. Data are the mean ± SD (n = 3, **p < 0.01; ***p < 0.001, Student’s t-test).
Loss of expression of either Kindlin-2, TβRI or ITGB1 inhibits signaling activities that are specific to β1-Integrin and TβRIIn the next set of experiments, we investigated the effect of loss of either Kindlin-2, TβRI or ITGB1 on oncogenic activities that are specifically activated downstream of either β1-Integrin or TβRI. For the β1-Integrin downstream activities, we assessed for cell adhesion on fibronectin, a cellular activity that is regulated by integrins [34] (Fig. 5A–D). Loss of expression of either of the three genes resulted in a significant (p < 0.001) inhibition of cell adhesion of MDA-MB-231 and 4T1 cells to Fibronectin (Fig. 5A, B and Fig. 5C, D, respectively). Adhesion of MDA-MB-231 and 4T1 cells to Matrigel (Fig. 5E–H, respectively) or Laminin (Fig. S6) was also significantly (p < 0.001) inhibited in the KO cells. Cell spreading on ECM is another major cellular activity that is tightly regulated by integrins [34]. Here again, we found loss of expression of either Kindlin-2, TβRI or ITGB1 resulted in a significant (p < 0.001) inhibition of spreading of MDA-MB-231 and 4T1 to Fibronectin (Fig. 5I–L, respectively), and to Matrigel (Fig. 5M, N and Fig. O&P, respectively), or to Laminin (Fig. S7). Thus we show loss of expression of either Kindlin-2, TβRI or ITGB1 to inhibits cellular activities that are specific to Integrins. Since β1-Integrin can form heterodimers with other α-Integrin subunits [35, 36], we used qt-RT-PCR to assess for expression levels of α1, αV and α5, which are predominantly expressed in cancer cells [37]. Loss of Kindlin-2 did not affect expression levels of either α1, αV and α5 subunits (Fig. S8A). However, loss of either ITGB1 or TβRI resulted in a significant (p < 0.01) inhibition of expression of both αV and α5 subunits, but not those of α1 subunit (Fig. S8B and S8C, respectively). This is consistent with the spreading data obtained on Fibronectin and Laminin since both αV and α5 integrin subunits, but not α1 subunit are involved in the β1-Integrin-mediated interaction with Fibronectin and Laminin [36]. Finally, as a readout for the molecular signaling downstream of TβRI, we assessed for phosphorylation levels of SMAD2/3 that takes place downstream of the TGF-β-mediated activation of the TβRI:TβRII complex. Loss of expression of either of the three proteins also resulted in reduction in phosphorylation levels of SMAD2/3 in both MDA-MB-231 (Fig. 5Q) and 4T1 cells (Fig. 5R). Thus, we show that loss of expression of either Kindlin-2, TβRI or ITGB1 inhibited the TβRI-specific downstream signaling, and, therefore, implicating Kindlin-2 as a major player in the regulation of the downstream signaling effectors of both β1-Integrin and TβRI.
Fig. 5: Loss of expression of either Kindlin-2, TβRI or ITGB1 inhibits signaling activities that are specific to β1-Integrin and TβRI.
Representative images and average number of adherent control (Ctrl) MDA-MB-231 and 4T1 cells, and their K2-KO, TβRI-KO and ITGB1-KO derivatives on fibronectin (A–D), and on Matrigel (E–H). I–N Representative images and average cell surface area of control (Ctrl) MDA-MB-231 and 4T1 cells, and their K2-KO, TβRI-KO and ITGB1-KO derivatives on fibronectin (I–L), and on Matrigel (M–P) after spreading assay. Actin filaments were stained with Alexa 488 Phalloidin and nuclei were counterstained with DAPI. Q, R, WB results of phosphorylation levels of SMAD2/3 in control (Ctrl) MDA-MB-231 and 4T1 cells, and their K2-KO, TβRI-KO and ITGB1-KO derivatives. Scale bar: 100 µm for adhesion images and 50 µm for spreading images. The numbers under each WB band represent the fold change in signal intensity with respect to its respective control band in each panel after normalization to the loading control signal. Data shown are representative of 3 replicates. Data are the mean ± SD (n = 3, **p < 0.01; ***p < 0.001, Student’s t-test).
Re-expression of Kindlin-2 in the K2-deficient TNBC cells is sufficient for the restoration of the oncogenic activities downstream of β1-Integrin and TβRITo determine whether the cellular activities that are regulated downstream of β1-Integrin are mediated by Kindlin-2, the K2-KO MDA-MB-231 and 4T1 TNBC cells re-expressing full-length Kindlin-2 were subjected to cell adhesion and spreading. Cell adhesion on fibronectin was fully restored in both K2-deficient MDA-MB-231 cells (Fig. 6A, B) and 4T1 cells (Fig. 6C, D) re-expressing Kindlin-2. Cell adhesion on Matrigel was also fully restored for both cell lines (Fig. 6E, F, and Fig. 6G, H), as well as Laminin (Fig. S9). Similarly, re-expression of full-length Kindlin-2 in the K2-KO cells also fully restored the spreading potential of both cell lines on fibronectin (Fig. 6I–L for MDA-MB-231 and 4T1, respectively), Matrigel (Fig. 6M–P for MDA-MB-231 and 4T1), and Laminin (Fig. S10). Phosphorylation levels of SMAD2/3, a readout of TβRI-specific downstream signaling activity, was also restored in K2-KO MDA-MB-231 cells treated with the proteosome inhibitor MG132 (Fig. 6Q), as well as in the K2-KO cells re-expressing full-length Kindlin-2 (Fig. 6R). Thus we confirm that the signaling activities that are mediated downstream of β1-Integrin or TβRI are specifically regulated by Kindlin-2. Additionally, we also confirmed that Kindlin-2 expression, by stabilizing the β1-Integrin:TβRI protein complex is sufficient for the restoration of these β1-Integrin and TβRI downstream cellular and signaling activities.
Fig. 6: Re-expression of Kindlin-2 in the K2-deficient TNBC cells is sufficient for the restoration of the oncogenic activities downstream of β1-Integrin and TβRI.
Representative images and average number of adherent MDA-MB-231 and 4T1 cells, and their K2-KO and K2-KO rescued with K2-full derivatives on Fibronectin (A–D), and on Matrigel (E–H). I–P Representative images and average cell surface area of MDA-MB-231 and 4T1 cells and their K2-KO and K2-KO rescued with K2-full derivatives after spreading assay on fibronectin (I–L) and on Matrigel (M–P). Actin filaments were stained with Alexa 488 Phalloidin and nuclei were counterstained with DAPI. WB analyses of phosphorylation od SMAD2/3 of control (Ctrl) MDA-MB-231 and 4T1 cells and their K2-KO derivatives after treatment with MG132 (Q) or after re-expression of full length in the K2-KO cells (R). Scale bar: 100 µm for adhesion images and 50 µm for spreading images. The numbers under each WB band represent the fold change in signal intensity with respect to its respective control band in each panel after normalization to the loading control signal. Data shown are representative of 3 replicates. Data are the mean ± SD (n = 3, ***p < 0.001, ANOVA).
Loss of expression of either Kindlin-2, TβRI or ITGB1 inhibits growth and metastasis of TNBC tumors, which can be restored by re-expression of Kindlin-2Next, we determined whether the biological and signaling effects observed in vitro, can also be recapitulated in vivo in mouse models for TNBC tumor progression and metastasis. Using the spontaneous metastasis mouse model, MDA-MB-231 or 4T1 TNBC control cells and their KO derivatives were injected in the mammary fat pads of NSG mice (MDA-MB-231) or Balb/C mice (4T1), and growth of the primary tumors was monitored over time. Loss of expression of either of the three proteins resulted in a significant (p < 0.001) delay in tumor growth and weight in both the MDA-MB-231 model (Fig. 7A, B) as well as the 4T1 model (Fig. 7C, D). Metastasis was also significantly (p < 0.001) inhibited in the lungs of mice injected with the KO MDA-MB-231 cells (Fig. 7E, F) or with KO 4T1 cells (Fig. G&H). Re-expression of Kindlin-2 in the Kindlin-2-deficient MDA-MB-231 cells resulted in the rescue of both tumor growth (Fig. 7I) and metastasis (Fig. 7J, K). These data show the impact of Kindlin-2, TβRI and ITGB1 on tumor progression and metastasis. They also confirm the specificity of Kindlin-2 in this process, where Kindlin-2 is sufficient for the restoration of the growth and metastasis potentials of TNBC tumors that lack expression of either TβRI or ITGB1.
Fig. 7: Loss of expression of either Kindlin-2, TβRI or ITGB1 inhibits growth and metastasis of TNBC tumors, which can be restored by re-expression of Kindlin-2.
Tumor volume (A, C) and weight (B, D) of NSG mice (A, B) or Balb/C mice (C, D) injected with control (Ctrl) MDA-MB-231 or 4T1 cells or their K2-KO, TβRI-KO and ITGB1-KO derivatives up to 40 days post injection. (Representative H&E staining (E, G) of lungs form tumor-bearing mice from A and C, and quantification of metastatic foci (F, H). I Tumor volume of NSG mice injected with control (Ctrl) MDA-MB-231 cells, and their K2-KO or K2-KO rescued with K2-full derivatives up to 36 days post injection. J Representative H&E staining of their corresponding lungs, and quantification of metastatic foci (K). **p < 0.01; ***p < 0.001; ANOVA.
Re-expression of Kindlin-2 lacking domains involved in the interaction of Kindlin-2 with β1-Integrin or TβRI does not fully restore the oncogenic behavior of TNBC tumorsFinally we asked whether re-introducing Kindlin-2 deletion variants that lack either the F2 or the F3 domain, that are required for the interaction with TβRI and β1-Integrin, respectively, would affect the oncogenic activities downstream of TβRI and/or β1-Integrin. We generated K2-KO-4T1 cell variants to express either GFP alone, K2-Full-GFP, ΔF2-K2-GFP, or ΔF3-K2-GFP (Fig. 8A). Western Blot analyses showed that re-expression of full-length Kindlin-2 in the K2-KO 4T1 cells to partially restore expression of both TβRI and β1-Integrin, as did the ΔF2, and ΔF3 Kindlin-2 variants (Fig. 8B). We also found that the re-expression of full-length Kindlin-2 partially restored colony formation (Fig. 8C, D), tumorsphere growth (Fig. 8E, F) and tumorsphere invasion (Fig. 8G, H), while re-expression of either ΔF2-K2 or ΔF3-K2 failed to restore these oncogenic activities since they remained close to those achieved in K2-KO group. Thus, these findings further support the requirement of these two domains, and therefore, the interaction of K2 with TβRI and β1-Integrin to maintain the oncogenic behavior of TNBC cells in vitro.
Fig. 8: Re-expression of Kindlin-2 lacking domains involved in the interaction of Kindlin-2 with β1-Integrin or TβRI does not fully restore the oncogenic behavior of TNBC tumors.
A Western Blot analysis with either Kindlin-2 or GFP antibodies showing expression of Kindlin-2-fusion derivatives from protein lysates of 4T1-K2-KO cells expressing either GFP, K2-Full-GFP, K2-ΔF2-GFP or K2-ΔF3-GFP. Note that the K2-ΔF2-GFP band is not detected by the Kindlin-2 antibody, since this antibody was raised against an epitope that resides within the F2 domain. B Western Blot analysis with indicated antibodies showing expressions of TβRI and β1-Integrin proteins from protein lysates of cells described in (A). β-Actin is a loading control in both A and B. 2D-colony formation assay (C, D), 3D-tumorsphere growth assay (E, F), and 3D-tumorsphere invasion assay (G, H) of 4T1-K2-KO cells, and their K2-Full, K2-ΔF2 or K2-ΔF3 rescued derivatives. Average number of adherent 4T1-K2-KO cells, and their K2-Full, K2-ΔF2 or K2-ΔF3 rescued derivatives on fibronectin (I), and Matrigel (J). K, L Average cell surface area of 4T1-K2-KO cells, and their K2-Full, K2-ΔF2 or K2-ΔF3 rescued derivatives. I, J Average number of spread 4T1-K2-KO cells, and their K2-Full, K2-ΔF2 or K2-ΔF3 rescued derivatives on fibronectin (K), and Matrigel (L). Scale bar: 100 µm for adhesion images and 50 µm for spreading images. M WB results of phosphorylation levels of SMAD2/3 4T1-K2-KO cells, and their K2-Full, K2-ΔF2 or K2-ΔF3 rescued derivatives. The numbers under each WB band represent the fold change in signal intensity with respect to its respective control band in each panel after normalization to the loading control signal. N Tumor volume of Balb/C mice injected with 4T1-K2-KO cells, and their K2-Full, K2-ΔF2 or K2-ΔF3 rescued derivatives up to 49 days post injection. Representative images of lungs (O) and quantification of metastatic foci (P) from mice described in (N). **p < 0.01; ***p < 0.001; ANOVA.
Next, we assessed for the effect of re-expression of ΔF2-K2 or ΔF3-K2 on the oncogenic activities downstream of either TβRI or β1-Integrin. Here again, we found that while K2-Full was able to restore the signaling activities downstream of β1-Integrin by restoring cell adhesion (Figs. 8I, J and S11) and cell spreading (Figs. 8K, L and S12) on different extracellular matrices, while neither ΔF2-K2 nor ΔF3-K2 were able to do so. Similarly, K2-Full was able to restore the signal transduction downstream of TβRI by increasing pSMAD expression levels as compared to K2-KO (Fig. 8M), while neither ΔF2-K2 nor ΔF3-K2 did. Thus, both the F2 and F3 domains, that are involved in the interaction of Kindlin-2 with TβRI and β1-Integrin, respectively, are required for the oncogenic activities of TNBC cells downstream of both TβRI and β1-Integrin.
Finally, we assessed for the effect of ΔF2-K2 and ΔF3-K2 on tumor growth and metastasis, and found that mice injected with K2-full-expressing K2-KO-4T1 cells showed tumor growth that is significantly (p > 0.001) higher than that in mice injected with K2-KO-4T1 cells (Fig. 8N). On the other hand, the tumors derived from mice injected with cells expressing either ΔF2-K2 and ΔF3-K2, although noticeably bigger than those derived from K2-KO cells, they were still significantly (p < 0.001) smaller than those derived from cells expressing full-length K2 (Fig. 8N). Metastatic dissemination to the lungs was also fully restored in mice injected with K2-Full-expressing K2-KO-4T1 cells, while ΔF2-K2- or ΔF3-K2-expressing K2-KO-4T1 cells only resulted in
留言 (0)