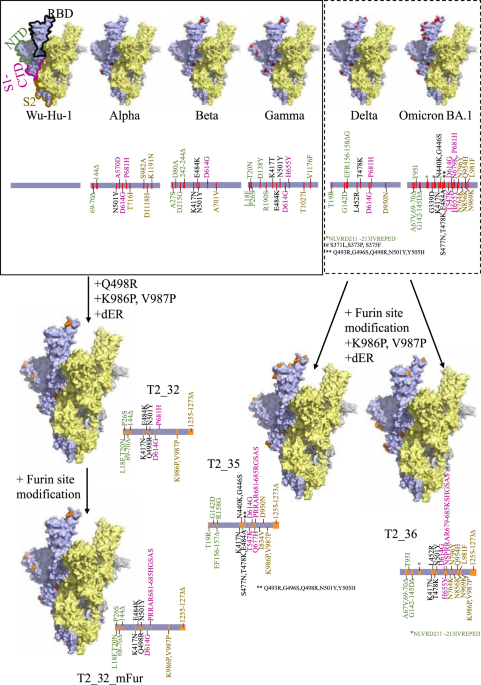
In-silico design of the vaccine antigens
A consensus sequence was generated for spike protein of VOCs - Alpha, Beta, and Gamma using the sequences deposited in NCBI Virus (Feb. 2021)34. Multiple sequence alignment (MSA) was generated for Wu-Hu-1 (NC_045512.2), Alpha, Beta, and Gamma using MAFFT algorithm35. The mutated amino acids for each VOCs were identified. Information on epitope residues were retrieved from the Immune Epitope DataBase (IEDB)14 and mapped along the length of the MSA to identify the mutations associated with immune inducing region of the spike protein. Further, epitope regions were classified as immunodominant region based on literature15. For further analyses, only mutations occurring in the immunodominant epitope region of the spike protein were considered, as they would contribute maximally to immune escape. Each mutation in immunodominant epitope were assigned to the four structural domains – N-terminal domain (NTD), receptor binding domain (RBD), C-terminal domain of S1 (S1-CTD), and stalk (S2) region and labelled with the VOCs it is observed in. The set of mutations from the NTD of the Alpha and Gamma variants, the set of mutations from the RBD region of the Beta, and the set of mutations from the S1-CTD of the Alpha variant were introduced on the backbone of the Wu-Hu-1 strain. Further mutations - K986P17,18, V987P17,18 and Q498R16 and deletion of 19 amino acids from C-Terminal of Spike protein were introduced to the final design -T2_32 (Fig. 1).
Using the similar design protocol, two designs – T2_35 and T2_36 were generated using MSA including the consensus Delta and Omicron BA.1 (Dec. 2021). Sets of mutations from the NTD and S2 domain of Delta variant and set of mutations from the RBD and S1-CTD of Omicron BA.1 were introduced on the backbone of Wu-Hu-1. In addition, mutations Q677H, I834V, K986P17,18, V987P17,18, replacing Furin cleavage site with GSAS motif17, and deletion of 19 amino acids from C-Terminal of Spike protein were introduced to the final design – T2_35 (Fig. 1). Sets of mutations from the NTD and S2 domain of Omicron BA.1 variant and set of mutations from the RBD and S1-CTD of Delta were introduced on the backbone of Wu-Hu-1. In addition, mutations K417N, K986P17,18, V987P17,18, replacing Furin cleavage site with GSAS motif17, and deletion of 19 amino acids from C-Terminal of Spike protein were introduced to the final design – T2_36 (Fig. 1).
The structural integrity of the resultant vaccine antigens was checked for by generating homology models using Modeller algorithm36. The 19 amino acids from the C-terminal of the spike protein were removed from all the wild-type controls as well. Subsequently, a Furin cleavage site modified version of T2_32 (referred as T2_32_mFur) was also designed.
Production and transformation of plasmids
Sequences of vaccine designs (T2_32 and Wu-Hu-1) were RNA- and codon-optimised for high level expression in human cells via the GeneOptimizer algorithm37. These genes were cloned into pEVAC38 (GeneArt/Thermofisher, Germany) via restriction digestion using kpn1 and not1. Plasmids were transformed via heat-shock in chemically competent E. coli DH5α cells (Invitrogen 18265-017). Plasmid DNA was extracted from transformed bacterial cultures via the Plasmid Mini Kit (Qiagen 12125). The DNA plasmids were purified using the EndoFree Plasmid Mega kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Plasmids were quality controlled by sanger sequencing and quantified using UV spectrophotometry (NanoDrop™ -Thermo Scientific) and assessed for absence of endotoxin.
Production of MVA
The MVA strain used in this study was MVA-CR1939. Recombinant MVA that expresses T2_32 was generated as described previously25. Briefly, for homologous recombination adherent CR.pIX cells were infected with MVA-CR19-GFP with different MOIs ranging from 0.5 to 0.006 and transfected with 0.4 μg shuttle plasmid pMVA Trans TK- SARS-CoV-2 T2_32 using Effectene (Qiagen, Germany). After 48 h the lysates were harvested, treated by three freeze-thaw cycles and sonicated. Then, different dilutions of the virus lysates were transferred to a fresh monolayer of AGE1.CR.pIX cells. After 2 h, a semisolid agarose overlay was applied and recombinant MVAs were identified by lacZ-staining using X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) and isolated with a pipette tip 72 h post infection. Genomic DNA was extracted from the isolated plaques after each agarose plaque purification round using the Quick-DNA Miniprep Kit from Zymo Research according to the manufacturer’s instructions. The correct integration into the TK locus was verified by PCR analysis using primers flanking the TK locus (CTCTCTAGCTACCACCGCAA and ATGCGTCCATAGTCCCGTTC). PCR products were separated on a 1% agarose gel, excised, and the expected sequence was confirmed by Sanger sequencing. The recombinant MVA-CR19 encoding T2_32 was plaque purified for additional three rounds and purified via two ultracentrifugation rounds at 20000 rpm at 4 °C using a 35% sucrose cushion.
The resulting recombinant MVA-CR19 T2_32 (MVA T2_32) virus stock was produced in suspension AGE1.CR.pIX cells as described previously40. Briefly, cells were seeded in shake flasks in a final volume of 45 mL in a mixture of CD-U7 (Xell) and DMEM (Gibco) (1:1) chemically-defined media, infected with an MOI of 0.1, and incubated in a shaking incubator with 180 rpm, 5 cm amplitude, 37 °C, and 5% CO2 (HT Multitron Cell, Infors AG, Switzerland). The culture was centrifuged 48 h post infection with 1100 × g for 5 min and 35 mL of the supernatant were discarded. The pellet was re-suspendend in 10 mL of the supernatant. This suspension was distributed into 10 vials and sonicated with a Vial Tweeter (Hielscher) for 20 s at 100% cycle and 90% amplitude.The virus titer in the lysate was determined on DF-1 cells using crystal violet staining. The absence of revertant or parental MVA and T2_32 insert identity was confirmed by PCR amplification and Sanger sequencing. Correct expression of T2_32 was confirmed by Western blot (40150-T62, Sino Biological; China) with cell lysates from HEK293 cells harvested 24 h after infection (MOI 2) with MVA T2_32.
Vaccination experiments in Guinea pigs
Two groups of four seven-week-old female Hartley guinea pigs were purchased from Envigo (Maastricht, Netherlands). Guinea pigs were immunised at 14 days intervals with 200 µg DNA vaccines bearing the antigen gene in the pEVAC vector, administered by intradermal route using the Pharmajet© device in a total volume of 200 µl over the hind legs. A third dose of DNA was administered at day 70 post prime and at day 112 was boosted intramuscular with MVA encoding T2_32 with 2.0E7 PFU/dose. Bleeds were taken through the saphenous vein and animals euthanised at the end of experiments using Pentoject under non-recovery anaesthesia.
Synthesis and packaging of mRNA
mRNA encoding the sequences of the vaccine antigens (T2_35, T2_36, T2_32_mFur, Wuhan-Hu-1, and Omicron BA.1) were synthesised by in vitro transcription (IVT) from linearised plasmid DNA templates using modified nucleotides to generate partial modified mRNAs. IVT was performed for 120 min at 37 °C using T7-RNA polymerase and co-transcriptional capping via Anti-Reverse Cap Analog (ARCA), followed by digestion of the template using DNAse I. After IVT, mRNAs were dephosphorylated for 15 min at 37 °C using alkaline phosphatase and enzymatically polyadenylated for 10-30 min at 37 °C using PolyA polymerase to produce a Poly A tail of approximately 120 nucleotides. Purification steps were performed by precipitation and subsequently formulated in water for injection at a concentration of 1 mg/mL. mRNAs were stored at -80 °C until LNP encapsulation. Each mRNA was LNP encapsulated via nanoprecipitation by microfluidic mixing of mRNA in citrate buffer (pH 4.5) with ionizable-, structural-, helper- and polyethylene glycol (PEG) lipids in ethanol, followed by buffer exchange and concentration via tangential flow filtration. mRNA LNPs were filtered through a 0.2μm membrane and stored at -20 °C until use. After manufacturing and freezing, the drug product was analytically characterised for particle size ( ≤ 100 nm), particle polydispersity ( ≤ 0.20), encapsulation efficiency ( ≥ 90%), mRNA integrity ( ≥ 90%) and mRNA identity (confirmed length).
Immunisation of mice
Female 8–10-week-old BALB/c mice (Charles River Laboratories, Kent, United Kingdom) were immunised twice at an interval of 21 days. A total volume of 100 µl of PBS containing 10 µg of lipid encapsulated mRNA encoding the antigens was administered intramuscularly over the two hind legs. The naïve mice group were administered 100 µl of vehicle only control. Bleeds were taken 3 weeks after each immunisation, and a final terminal bleed 6 weeks after the second immunisation under non-recovery anaesthesia.
Production of lentiviral pseudotypes
Lentiviral pseudotypes were produced by transient transfection of HEK293T/17 cells with packaging plasmids p8.9141,42 and pCSFLW43 and different SARS-CoV-2 VOC spike-bearing expression plasmids in the pEVAC backbone, using the Fugene-HD (Promega E2311) transfection reagent44,45. Supernatants were harvested after 48 h, passed through a 0.45 µm cellulose acetate filter, and titrated on HEK293T/17 cells transiently expressing human ACE-2 and TMPRSS2. Target HEK293T/17 cells were transfected 24 h prior with 2 µg pCAGGS-huACE-2 and 150 ng pCAGGS-TMPRSS2 in a T75 tissue culture flask46,47. The sequences of the VOCs spike used for the production of pseudotypes are provided in the supplementary file.
Pseudotype-based micro-neutralisation assay
Pseudotype-based micro-neutralisation assays (pMN) were performed as described previously48. Briefly, serial dilutions of serum were incubated with lentiviral pseudotypes bearing SARS-CoV-2, and SARS-CoV-2 VOC spikes for 1 h at 37 °C, 5% CO2 in 96-well white cell culture plates. 1.5 × 104 HEK293T/17 cells transiently expressing human ACE-2 and TMPRSS2 were then added per well and plates incubated for 48 h at 37 °C, 5% CO2 in a humidified incubator. Bright-Glo (Promega) was then added to each well and luminescence read after a five-minute incubation period. Experimental data points were normalised to 100% and 0% neutralisation using cell only and virus + cell control wells respectively and non-linear regression analysis performed in GraphPad Prism 9 to produce neutralisation curves and IC50 values.
Statistical analyses
Two-tailed Mann–Whitney U tests were performed for all the pairwise comparisons using the Python sklearn package49. All the plots were generated using the Python Matplotlib package and statannotat package50.

留言 (0)