Source materials
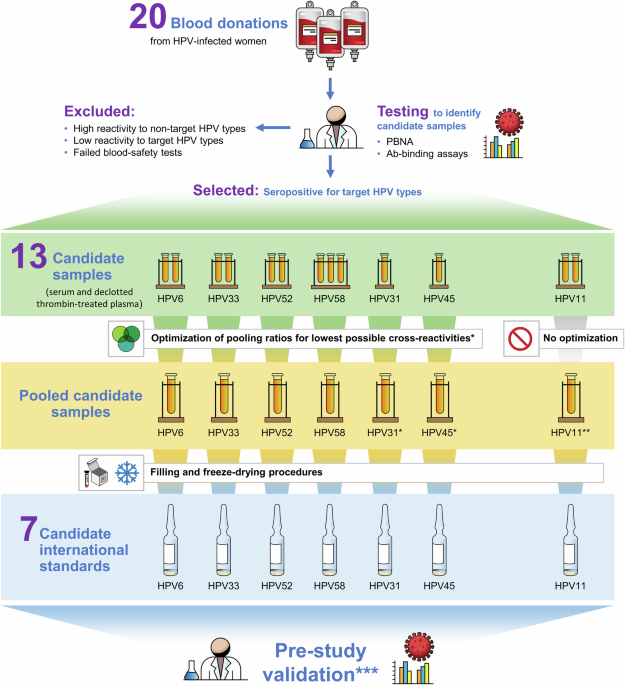
The process for testing, selection, and formulation of the source materials to produce the 7 candidate standards is shown in Fig. 1. Anonymized serum and plasma samples were provided to NIBSC for development into candidate materials. Donations obtained by informed consent from women naturally infected with HPV were provided by Professor Joakim Dillner, Karolinska Institute, Stockholm, Sweden, in collaboration with Dr. Jarunya Ngamkham, National Cancer Institute, Thailand (approved by the ethical and research committees of the National Cancer Institute (NCI), Thailand (EC 122/2009, decision taken 18.12.2009)21; Professor Mario Poljak, University of Ljubljana, Ljubljana, Slovenia (approved by the Medical Ethics Committee of the Republic of Slovenia (Consent number: 83/11/09) and also approved by the Ethical Committe of Umeå, Sweden (Nr. 118/92, 95-2400 and 98/12)22; and Dr. Weijin Huang, National Institutes for Food and Drug Control Beijing, P.R. China by which samples were selected from a large number of anonymized samples from a plasma center (Shanghai RAAS Blood Products Co. Ltd, Shanghai, China)23. Prior to development, the plasma donations were converted to serum by thrombin-treatment to cause clotting followed by defibrination by centrifugation and then filtering the supernatant. Twenty donations were initially tested by 4 external HPV reference laboratories (Supplementary Table 13) in validated PBNA and Ab-binding assays to identify the materials most suitable for development into candidate standards. Preselection criteria for development included: i) confirmation of seropositivity (as defined by each reference laboratory for their assay) to the HPV type of interest and no reactivity to other HPV vaccine types i.e., each candidate IS should be monospecific to allow assignment of clearly defined IU that is not affected by the presence of possibly cross-reactive antibodies against other HPV types.; ii) each candidate should consist of donations from at least 2 donors in order to reduce the risk that a donation with uncommon characteristics would be selected as an IS; iii) donations should be obtained from only women as data suggest that women have higher antibody responses to natural infection than men24.
The initial testing round identified 13 donations for development into candidate standards; however, exceptions to the selection criteria were made for the candidate antibody standards for HPV31, HPV45, and HPV11 due to the difficulty in sourcing monospecific source materials. In the case of the HPV31 and HPV45 candidate standards, only single donations were identified for development. For the candidate standard for HPV11 antibodies, only 2 donations were available for development. These were shown also to be reactive to HPV6. One of the donations was also reactive to HPV33, 52 and 58 across 1 or more methods. The cross reactivity of the HPV11 materials can be attributed, in part, to cross-reactive epitopes with other HPV types14,25 and/or co-infection. The use of the 2 available donations was taken as a best-case scenario for the formulation of the candidate HPV11 antibody standard.
Initial testing found low-level reactivity to at least 1 non-target HPV type in at least 1 donation for each candidate. A small-scale mixing study was performed to identify the pooling ratio for each candidate standard to mitigate the low-level, non-target reactivity while maintaining optimal seroreactivity for the target HPV type (Fig. 1). In the case of the HPV31 and HPV45 candidates, pooling was performed using HPV antibody-negative serum (Table 1, Sample E). The optimum pooling ratios for formulations were determined in a small-volume mixing study for candidate antibody standards HPV6, 31, 33, 45, 52, and 58 tested by 2 HPV reference laboratories (REF-1, REF-2 in Supplementary Tables 19−23). The HPV11 candidate standard was formulated by mixing the 2 donations without optimization.
Production and pre-study testing of candidate International Standards
The NIBSC Human Materials Advisory Committee approved the use of the source materials for development into candidate standards (approval reference 18/07/DW). All donations used to make the candidates were tested at NIBSC and found negative for Hepatitis B surface antigen (HBsAg), antibodies to Human Immunodeficiency Virus (HIV) 1/2, and Hepatitis C (HCV) RNA. From March 2020 to June 2021, NIBSC undertook separate productions for each candidate standard. Materials were filtered, pooled and dispensed at high precision in 250 µL aliquots into glass ampoules and freeze-dried according to standard operating procedures and WHO guidelines6. The ampoules were sealed under 1 atmosphere of nitrogen and stored at −20 °C. Ampoule seal integrity was assessed by measuring residual oxygen by frequency modulation spectroscopy (FMS-760 from Lighthouse Instruments, Charlottesville, VA, USA) using a laser infra-red source beamed through the headspace of the ampoule. Residual moisture content was measured using the colorimetric Karl Fischer method (Mitsubishi CA-100 or CA-200, and kit obtained through A1- Envirosciences, Blyth, UK). Each candidate was assigned a unique NIBSC product code. Descriptions of the candidate ISs, including the anonymous donor identifiers, pooling information, manufacture, and validation outcomes, are provided in Supplementary Table 24. An anomaly occurred during the production of the HPV11 candidate where the material was dehydrated rather than lyophilized due to a freeze-drier failure although validation testing showed that the reactivities of all the candidate standards were similar to the results obtained during the selection process. In summary, the pre-study results confirmed the suitability of the 7 candidates for formal evaluation in the international collaborative study (Supplementary Tables 19−23).
Sample panel
The samples distributed for testing in the collaborative study are listed in Table 1. To demonstrate that the candidate standards would be suitable for use in assays developed to monitor antibody responses in sera from individuals naturally infected with HPV as well as those vaccinated with different HPV vaccines (i.e. aspects of commutability), coded samples of each type of sample were distributed with the candidates. The HPV antibody-negative serum pool used in the mixing study was included as a negative control. HPV antibody-negative serum and sera from recipients of the 9v vaccine, were collected by Occupational Health Services at the National Institutes of Health (NIH) National Cancer Institute (NCI) at Fort Detrick, MD, under the Research Donor Protocol (RDP). Participants were healthy NCI-Frederick employees and other NIH staff that donated blood samples for in vitro research at the NCI-Frederick laboratories. The protocol is listed under NIH protocol number OH99CN046 and NCT number NCT00339911. These OHS samples were used for reagent optimization.
Pooled plasma from recipients of the 2v vaccine were provided by Dr. Simon Beddows, UK Health Security Agency, Virus Reference Department, London, UK Plasma packs were obtained from NHS Blood and Transplant, UK, Formal approval was sought from the NHS Blood and Transplant according to their own release procedures for samples considered for non-clinical use. No individual identifying information was available and no additional individual consent was required14.
Study samples were delivered on dry ice to participating laboratories, who were instructed to store these materials at or below -20°C.
Study protocol
Participants were requested to use their established method(s) for the detection and quantification of antibodies to 1 or more of the 9 HPV types (HPV6, 11, 16, 18, 31, 33, 45, 52, 58). For each HPV type tested, participants were requested to test all study samples concurrently in 3 independent assays using a new set of prepared samples for each run. Participants were provided with an extra set of samples to allow for a preliminary assay run to determine optimal dilution ranges for testing. Participants were requested to set up and test at least 2 independent replicate series of dilutions (NOT 2 aliquots from a single dilution series), where practical. Written instructions and a web conference were provided to guide participants on the preparation and testing of the study samples including a recommended plate setup to ensure that certain unknown samples were assayed together to allow direct comparability.
Participants were requested to record the readout (e.g., +/−, optical density [OD], relative light units [RLU]) for each dilution and include the cut-off value indicating sero-reactivity for each assay, stating how the cut-off criteria was established and whether each sample dilution tested was considered positive or negative according to assay criteria. Participants were also requested to provide information on the preparation and use of VLPs/capsids/pseudovirions in their assay(s).
Participants
NIBSC invited 29 HPV laboratories worldwide to participate in the collaborative study. A draft protocol was provided to the prospective participants for their review and comment. Twelve laboratories accepted the invitation with 1 subsequently withdrawing from the study. Laboratories that participated in the study are referred to by code numbers (Supplementary Table 1) allocated at random and not representing the order of listing at the end of the paper.
Collaborative study assay methods
The assay methods performed by participants fall into 2 general categories: PBNA and Ab-binding assays. None of the participants shared a common standard operating procedure or protocol for performing the assay. The specific details of the individual assay methods used by participating laboratories are not described here to ensure the coding of the participating laboratory results remain anonymized. In general, participants performing PBNA used PsV that were prepared in-house using 293FT, 293TT or HeLaT cells. Reporter genes for measuring neutralization included those encoding for green fluorescent protein (GFP), red fluorescent protein (RFP), cyan fluorescent protein (CFP), secreted alkaline phosphatase (SEAP) and (nano)luciferase. Reporter gene readouts included fluorescence, fluorospots, OD, chemiluminescence, and RLU. PBNA cut-off values for seropositivity were defined based on the lowest dilution tested or in reference to a non-relevant PsV. Initial dilutions ranged from 1/10 to 1/100 with serial dilution steps ranging from 2-fold to 5-fold. Results were reported in neutralizing titers. Two laboratories also reported results in IU/mL traceable to the established ISs for HPV16 and HPV18 antibodies.
Participants performing Ab-binding assays described the antigen used as type-specific VLP, L1L2 VLP, pseudovirion or “antigen” with no further detail. Methods of antigen production included mammalian cell, Pichia pastoris and Escherichia coli expression systems. Readouts for Ab-binding assays included OD, mean fluorescence intensity (MFI) and RLU. Initial dilutions ranged from 1/50 to 1/100 with serial dilution steps ranging from 2-fold to 10-fold. Four laboratories used the readouts to calculate antibody-binding levels in laboratory-defined units relative to an in-house standard. In the case of HPV16 and HPV18, the same 4 laboratories reported in IU/mL. Four laboratories reported their cut-off values for sample seropositivity with 2 laboratories reporting that the cut-off values were determined using sera from children. Two laboratories reported endpoint titers with no cut-off defined for seropositivity.
Statistical methods
Statistical analysis of assay results within and between laboratories is based on the general principles for establishing International Standards and other biological reference materials for serology6,26,27,28.
Samples were scored as positive ‘P’ or negative ‘N’ for antibodies based on the criteria defined by the participating laboratories. For PBNA, if no cut-off for seropositivity was provided, a sample was scored negative if the titer was less than the lowest dilution tested. For Ab-binding assays, samples were not scored if the cut-off was not defined. An overall laboratory score was assigned to each sample according to the majority response reported across independent assays (e.g., 2 out of 3 assays). Data was assessed further if the sample scored seropositive by > 50% of laboratories. At this point, the Ab-binding levels reported by the 2 laboratories with no defined cutoffs were included in the analysis.
Overall antibody concentrations, i.e., titers or “units”/mL, for each sample were calculated as the median across the independent assays for each laboratory, method, and HPV type.
Intra-assay variability was assessed by calculating the ratios of median antibody concentration for duplicate samples I & P, A & J, B & K (Table 1). A ratio of 1 indicated matching results. Ratios less than 0.8 or greater than 1.20 (i.e., more than 20% difference in measurements) were taken as higher variability. A ratio of 0 indicated that 1 of the duplicate samples was scored negative by the laboratory. Intra-assay variability of results for antibodies against HPV6 and 11 (and 58 in PBNA only) was not assessed due to the limited availability of seropositive samples for inclusion in the study.
Inter-assay variability within each laboratory was assessed by calculating the ratio of the maximum and minimum antibody concentration (un-transformed) reported for each sample across independent assays. A > 4-fold difference in the maximum and minimum concentrations (Max:Min) was selected as an indicator of greater intra-laboratory variability.
Overall mean results across laboratories were calculated for each sample as the GM of the laboratory median antibody concentrations or potencies expressed relative to the indicated candidate standard. For the purpose of this assessment, candidate standards were assigned a value of 1 to determine overall GM relative potencies. The between laboratory (inter-laboratory) variability was assessed by calculating GCV using the equation GCV= [10s-1] × 100% where s is the standard deviation of the log10 transformed median values. Ratios of the maximum and minimum results (un-transformed) across laboratories were also calculated.
Thermal stability assessment
An accelerated thermal degradation (ATD) study was performed in order to predict the long-term stability of the candidate standards when stored at the recommended temperature of −20 °C. Ampoules of the candidate standards were stored at −70 °C, −20 °C, 4 °C, 20 °C, 37 °C and 45 °C and then removed at indicated time points and held at −70 °C or below until assayed. The ATD samples were shipped on dry ice to 2 HPV reference laboratories (REF-1 and REF-2) for testing in PBNA and Ab-binding assays. The ATD samples were tested concurrently against the respective baseline sample (−70 °C or −20 °C) in at least 3 independent assays. Estimates of antibody concentration of the ATD samples (titers for PBNA, units/ml for Ab-binding) were reported to NIBSC for analysis. GM potencies and 95% confidence limits for the ATD samples stored at the elevated temperatures were calculated relative to the material stored at the baseline temperature (−70 °C or −20 °C). The baseline samples were assigned a value of 1 for the purpose of the assessment. To predict the degradation rate of the candidates when stored at −20 °C, the relative potencies of the ATD samples were used to fit an Arrhenius equation assuming first-order decay15.

留言 (0)