This study elucidates a novel mechanism to explain the underlying pathophysiology of lung inflammation in chronic airway diseases characterized by increased TGF-β1 signaling. Specifically, we show that TGF-β1 upregulates miR-449b-5p, which directly suppresses LPO. This is the first report of microRNA-mediated regulation of LPO. TGF-β1-mediated LPO suppression leads to increased ASL H2O2 levels and a consequent increase in proinflammatory cytokines.
Cell types
Primary Normal human bronchial epithelial (NHBE) cells and Human Bronchial Epithelial Cell Line (BEAS-2B) were used in these studies. NHBE were obtained from the University Of Miami Life Alliance Organ Recovery Agency (LAORA) and re-differentiated at the Air-Liquid Interface (ALI) as described by Dr. Fulcher and colleagues, Dr. Nlend and colleagues and adopted by us [20, 22,23,24]. The primary cultures undergo mucociliary differentiation at the ALI, reproducing the in vivo morphology and key physiologic processes to regenerate the native bronchial epithelium ex vivo [20, 22,23,24]. Experiments used cells from the lungs of non-smoker donors that were negative for HIV, Epstein-Barr virus (EBV), Cytomegalovirus (CMV), and Hepatitis B (HBV) not to confound the findings in unknown ways. Lung donors from both sexes were used.
BEAS-2B bronchial epithelial cell lines were obtained from the ATCC (Rockville, MD, USA) and maintained in Bronchial Epithelial Cell Growth Medium (BEGM) as previously described by us [20, 22].
Primary cells from lung tissues of healthy non-smokers (control) and COPD patients were purchased from BioIVT (Westbury, NY) to analyze and compare TGF-β1 and LPO expression.
10ng/ml TGF-β1 treatment in NHBE and BEAS-2B
Recombinant TGF-β1 (R&D Systems, Cat # 240-B-002) was dissolved according to the manufacturer’s instructions at a 10 μg/μL stock concentration. The final concentration for all treatments was 10 ng/mL in ALI (NHBE cells) media or BEGM (BEAS-2B cells). For NHBE cells, ALI media containing TGF-β1 or media only (control) were added basolaterally (1mL) and apically (100 μL) to mimic physiological conditions. For BEAS-2B, 10ng/mL was directly added to the cells for the treatment. The 10ng/ml treatment of TGF-β1 was used within the mean physiological range (2–20 ng/mL) established by Dr. Sun and colleagues and adopted by us [20, 25].
25 μM aurintricarboxylic acid (ATA) treatment in NHBE ALI cultures
ATA (Sigma-Aldrich, Cat # A1895-5G) was dissolved according to the manufacturer’s instructions at a stock concentration of 25mM. NHBE ALI cultures grown on snap-wells and were treated with 10ng/ml of TGF-β1, and separately, a second set was treated with 25μM of ATA 3 hours prior to the TGF-β1 treatments. After 24 hours, RNA was collected and purified to analyze the expression of LPO.
Cigarette smoke exposure
NHBE ALI and BEAS-2B cultures were exposed to air (control) or cigarette smoke using the SCIREQ smoke robot (Montreal, QC, Canada). Four cigarettes of research-grade (Research cigarette, University of Kentucky College of Agriculture: Cigarette Program: Code: 3R4F) were smoked, with a puff volume of 35 mL for 2 seconds every 30 seconds and blown over cell culture at a rate of 5 mL/minutes, according to the International Organization for Standardization (ISO) 3308. Control cells were similarly exposed to air (air control). We have shown that the smoke regimen does not affect the integrity of the epithelium or cell viability [8].
Quantification of mRNA expression by quantitative reverse transcription-PCR (qRT-PCR)
Using the website protocol, RNAs were isolated and purified from NHBE and BAES-2B cells treated/exposed to CS or transfected with different treatments using the PureLinkTM RNA Mini kit (Invitrogen, Cat # 12183025).
After the RNA was purified, its purity and concentration were analyzed by O.D. 260/280 nm absorbance ratio higher than 2.0 by the Synergy HT Multi-Mode Microplate Reader from BioTek, Winooski, VT, USA. 500ng of total RNA was reverse transcribed (RT) to synthesize cDNA using a high-capacity cDNA reverse transcription kit (ThermoFisher Scientific, Cat # 4368813).
Then, the relative amount of mRNA was quantified using TaqMan Fast Advanced Master Mix kit (ThermoFisher Scientific, Cat # 4444557) in 20 μL real-time PCR reactions for human LPO probe (ThermoFisher Scientific, Cat # HS00976392), mouse LPO probe (ThermoFisher Scientific, Cat # Mm00475466) and human TGF-β1 probe (ThermoFisher Scientific, Cat # HS00998133) using the BioRad CFX96 real-time system. The following parameters for reverse transcription were applied for 10 min at 25 °C, 30 min at 37 °C 2X, 30 min at 37 °C, and 5 min at 85 °C, followed by a holding step at 4 °C.
All data were normalized to human GAPDH (ThermoFisher Scientific, Cat # Hs02786624) or mouse GAPDH (ThermoFisher Scientific, Cat #Mm 99999915) according to the samples’ species and human 18S probe (ThermoFisher Scientific, Cat # HS99999901) and calculated as mean fold change in expression of the target gene using the comparative CT method.
For the expression miR-449b-5p, the RNA was purified and quantified as described above, then 10ng of total RNA was reverse transcribed (RT) using the TaqMan MicroRNA Reverse transcriptase kit (ThermoFisher Scientific Cat # 4366597) and the 5X Probe for MiR-449b-5p (ThermoFisher Scientific, Cat # RT001608). Then, the relative amount of mRNA was quantified using TaqMan Fast Advanced Master Mix (kit (ThermoFisher Scientific, Cat # 4444557) in 15 μL real-time PCR reactions for miR-449b-5p probe (ThermoFisher Scientific, Cat # HS06626452), using the BioRad CFX96 real-time system. The following parameters for reverse transcription were applied for 30 min at 16 °C, 30 min at 42 °C, and 5 min at 85 °C, followed by a holding step at 4 °C.
Quantification of protein expression by a Western blot
Proteins were collected using a solution of RIPA buffer (ThermoFisher Scientific, Cat # 89901) with a tablet of Protease inhibitor cocktail (Sigma-Aldrich, Cat # 11836153001). After treatment, cells were washed with PBS buffer pH 7.4 (Fisher Scientific, Cat # 2306394) and 400ul of RIPA/Buffer plus protease inhibitor tables were added. Then, the samples were placed in a slow speed shaker for 15 minutes followed by the collection, sonication (frequency of 20 kHz, with 120 Watts gently moving the tube up and down 3 times every 10 seconds) and centrifugation (14000 x g for 15 min) of the samples. Proteins were quantified by Bio-Rad Protein Assay Dye (Bio-Rad, Cat # 500006) and the Spectrophotometer Genesys 10 (Thermo Scientific) according to the manufacturer’s instructions.
Western Blot was performed using the Bio-Rad Western blot kit with 4–20% precast gel (Bio-Rad, Cat # 4568094) and using dual color Precision Plus Protein Standards (Bio-Rad, Cat # 161–0374).
After proteins were quantified, 50ng of total proteins were loaded onto a gel and run at 60 V and 80 V, respectively, for 1 hour and 45 min. Proteins were transferred to the PVDF membrane at 100 V for 1 hour. Following the transfer, Blot was blocked in 10% Blocking-Grade Blocker (Bio-Rad Cat # 1706404) in TBS-Tween 20 (TBS: Bio-Rad, Cat # 1706435 and Tween 20: Sigma Cat # 9005-64-5) for an hour and then incubated in 5% Blocking-Grade Blocker in TBS-Tween 20 with primary antibody for LPO (1: 1000; Sigma-Aldrich Cat # SAB2500580) overnight. The next day, the Blot was washed four times and incubated for an hour with an anti-goat second antibody diluted to a concentration of 1:2500 in 1% Blocking-Grade Blocker in TBS-Tween 20. Next, the Blot was washed four times, and bands were detected in the Bio-Rad Chemidoc Imaging system (Bio-Rad, Cat # 12003153) using the Thermo Scientific™ SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Scientific, Cat # PI34095). The same day, the Blot was stripped using Restore Plus Western Blot Stripping buffer (Thermo Scientific Cat # 46430) and re-probe for GAPDH (1:5000; Sigma-Aldrich, Cat # G9545) for normalization. The relative density of the detected protein bands was measured using the ImageLab Ink software, and the values obtained were averaged. Each samples were run in triplicate.
H2O2 quantitation by Amplex Red Assay
NHBE ALI cultures were treated with TGF-β1 (10ng/ml) for 24 hours and exposed to cigarette smoke for 48 hours. Then, according to the manufacturer’s instructions, H2O2 levels (uM) in the ASL wash were quantified using the Amplex Red Assay kit (Invitrogen, Cat # A22188).
Staining by C12FDG dye to estimate senescence
The senescent marker SA-β-Gal was analyzed by staining with C12FDG (ThermoFisher Scientific, Cat # D2893), Bafilomycin A1 (Sigma Aldrich Cat # B1793), and counterstaining with DAPI (Southern Biotech, Cat # 0100 − 20) as a nuclear dye. In four-chamber slides (Thermo Scientific Cat # 177399), BEAS-2B cells were treated with 10ng/ml of TGF-β1 (R&D system, Cat # 240-B) (Set 1) and exposed to cigarette smoke (Set 2) individually. The media was substituted with fresh medium with respective treatments every 48 h. Six days post-treatment, cells were stained using 33 μM C12FDG. The cells were first washed with PBS buffer pH 7.4 and treated with 50nM Bafilomycin A1 following an incubation time of 1 h at 37 °C. Next, cells were washed with PBS and treated with 33μM of C12FDG following an incubation time of 1 hour and 30 minutes at 37 °C. Next, the chamber separator was removed, and cells were washed twice in PBS for periods of 10 minutes, followed by fixation with 4% PFA in PBS (ThermoFisher Scientific Cat # J19943.K2) for 15 minutes. After 15 minutes of fixation, cells were washed with PBS for 10 minutes, and ∼ 30uL of DAPI was added to the slides covered with a coverslip and kept outside for drying (protected from light). Images were acquired using a Keyence All-in-One microscope (40X) at the same exposure time; Scale bars 50 μm. Mean intensity was quantified using the ImageJ browser for the National Institutes of Health (NIH). Experiments were performed in triplicates of each set.
The same staining procedures were also performed in a third set of BEAS-2B transfected with 40nM of miR-449b-5p mimic. Briefly, in four-chamber slides, BEAS-2B were transfected with 40nM of miR-449b-5p mimic using the RNAiMax reagent (ThermoFisher Scientific Cat # 13778-030) following the manufacturer’s instructions. After 48 hours post-transfection, cells were stained using C12FDG. Experiments were performed in triplicates of each set.
Mitophagy staining by MitoTracker Green to detect defective mitophagy
MitoTracker Green (ThermoFisher Scientific, Cat # M7514) was used to estimate and analyze the mitochondria mass to check for defective mitophagy. The MitoTracker Green FM probe is non-fluorescent in aqueous solutions and only becomes fluorescent once it accumulates in the lipid environment inside the mitochondria. Since the dyes passively diffuse across the plasma membrane and accumulate in mitochondria, MitoTracker Green was used in a concentration of 100nM and was counterstained with DAPI. Identical treatments of the 3 sets of BEAS-2B stained with C12FDG were stained to MitoTracker Green. After six days (sets 1 and 2) and 48 hours (set 3) post-treatment, cells were stained using MitoTracker Green. First, the cells were washed with PBS buffer pH 7.4 and treated with 100nM MitoTracker Green following an incubation time of 20 minutes at 37 °C. Next, the chambers separator was removed, and cells were washed twice in PBS for periods of 10 minutes, followed by the counterstaining with DAPI ∼ 30 uL and covered with a coverslip until dry (∼ 48 hours). Images were acquired using a Keyence All-in-One microscope (100X) at the same exposure; Scale bars 10 μm. Mean intensity was quantified using the ImageJ browser from the National Institutes of Health (NIH) website. Experiments were performed in triplicates of each set.
The same staining procedures were also performed in a third set of BEAS-2B transfected with 40nM of miR-449b-5p mimic. Briefly, in four-chamber slides, BEAS-2B were transfected with 40nM of miR-449b-5p mimic using the RNAiMax reagent (ThermoFisher Scientific Cat # 13778-030) following the manufacturer’s instructions. After 48 hours post-transfection, cells were stained using MitoTracker Green. Experiments were performed in triplicates of each set.
MicroRNA sequences & magnetofection
Hsa-miR-449b-5p mimic (Sequence: AGGCAGUGUAUUGUUAGCUGGC) and miRNA inhibitor (antagomir-449b-5p) (Product ID: MH11521) were purchased from Sigma. Magnetofection was performed to transfect NHBE cells with 40nM of miR-449b-5p mimic, 10ng/mL of TGF-β1, and antagomir + TGF-β1 using the PolyMAg reagent (Boca Scientific, Cat # PN30200) following manufacturer’s instructions.
RNAiMax transfection
For experiments involving staining for mitophagy and senescence, BEAS-2B cells were transfected with 40nM of miR-449b-5p mimic using the RNAiMax reagent (Invitrogen, Cat #13778-030) according to the manufacturer’s directions.
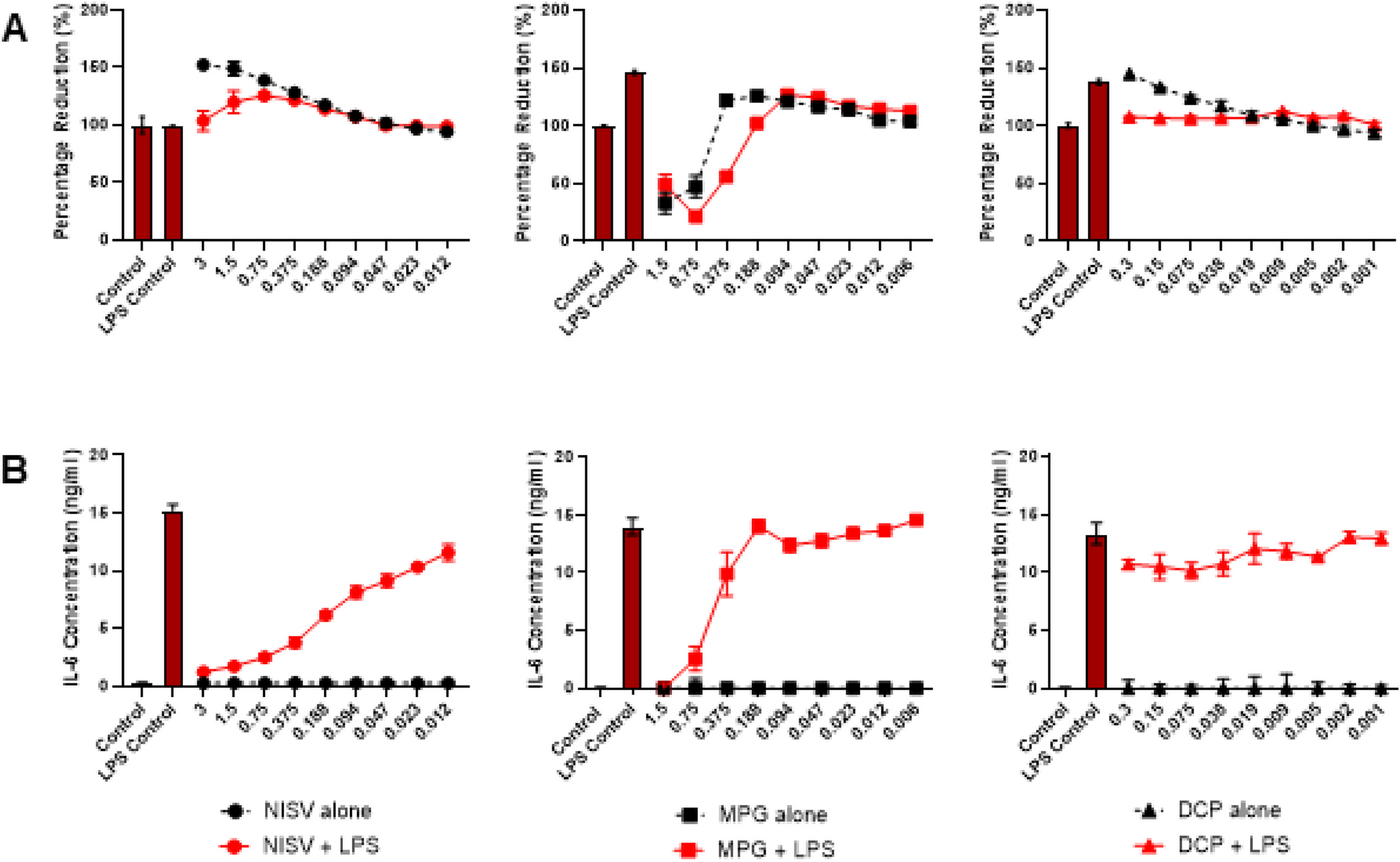
Pro-inflammatory mediators by Luminex
NHBE cells were treated with TGF-β1 (10ng/ml); media was substituted with fresh medium with the treatment every 48 hours for six days. On day 6th, culture supernatants were collected and analyzed by Luminex multiplex assay (Bio-Plex Pro human cytokine immunoassay kit (Bio-Rad. Cat # M500KCAF0Y) as we have described [26]. Experiments were performed in triplicate from 3 different lung samples. Only values higher than 2 fold were taken into consideration for representation. * Significant (p < 0.05).
Statistical analyses
The data were analyzed using unpaired t-tests for two groups or ANOVA followed by Tukey Kramer’s honestly significant difference test for multiple comparisons as appropriate. The significance was considered at the level of p < 0.05 for all the data. All tests were analyzed using GraphPad Prism software (version 9.5.1).




留言 (0)