Experimental animals
The Experimental Animal Center of Zhejiang Academy of Medical Sciences (License No. SYXK (Zhe) 2019-0001) supplied six-week-old male SD rats for the study. The animal experiments were carried out in compliance with the National Research Council’s Guide for the Care and Use of Laboratory Animals. The welfare of animals and the experimental procedures were approved by the Institutional Animal Care and Use Committee of Zhejiang Chinese Medical University (Hangzhou, China) (ZSLL-2017-040) in 2017. All rats were acclimatized and fed for one week, their basal blood pressure was measured and randomized into 4 groups according to their basal blood pressure, which were the normal group (NG, n = 10), the model group (MG, n = 10), the BUDE low-dose group (BUDE-L, 75 mg·kg− 1, n = 10), and the BUDE high-dose group (BUDE-H, 150 mg·kg− 1, n = 10). All rats except normal rats were fed high-fat chow (15% lard, 0.8% cholesterol, 20% sucrose, 0.2% sodium cholate, and 64% regular chow; protein: fat: carbohydrate = 19:37:44), as well as compound gradient drinking. During the modeling process, the rats in the drug administration group were given a daily dose of the designated test drug, while the normal and model groups were given water gavages for a duration of six weeks. All rats were then euthanized with an intraperitoneal injection of 0.2 g/mL urethane at a volume of 0.6 mL/100 g, ensuring the animals were relieved of stress and discomfort while maintaining a stable physiological state.
Cells and cell culture
Mouse monocyte macrophage leukemia cells (RAW264.7), derived from laboratory reserve, were cultured in DMEM (Gibco, USA) medium supplemented with 100 U/mL streptomycin-penicillin (Biosharp, China) and 10% fetal bovine serum (Four Seasons, China) at 37 °C in 5% CO2. Cells in logarithmic growth phase were then inoculated in 6-well plates with a cell suspension of 1 × 106 cells/well (2 mL per well). After 24 h of plate laying, a blank control group, a model control group, and three BUDE groups with different concentrations (25, 15, and 5 µM) were established. One hour after drug administration, while the other groups were given lipopolysaccharide (LPS) at 200 ng/mL to simulate the inflammatory response, the blank control group was given the same amount of baseline media. The drug action was continued for 6 hours for further subsequent experiments.
Tested drugs
Chrysanthemum indicum L. was acquired from Zhejiang Chinese Medical University, Traditional Chinese Medicine Decoction Pieces Co., Ltd. (Hangzhou, Zhejiang, China, LOT: 2,001,043). Its identification can be referenced in the 2020 Chinese Pharmacopoeia. The drug used in this study was derived from BUDE that had been extracted in a previous study. In the previous study, the results of HPLC analyses demonstrated that BUDE contained 69.62 ± 0.78% of BUD [23]. The required BUDE for rats were obtained by weighing the dry powder of BUDE and configuring it with pure water. The dose for rats in this study was derived from the adult dosage in the Chinese Pharmacopoeia and obtained by converting the surface area of the human body. The specific dose was mainly referred to the results of previous studies. For cells, BUDE prepared by dissolving BUDE powder in DMSO (Shanghai yuanye, China).
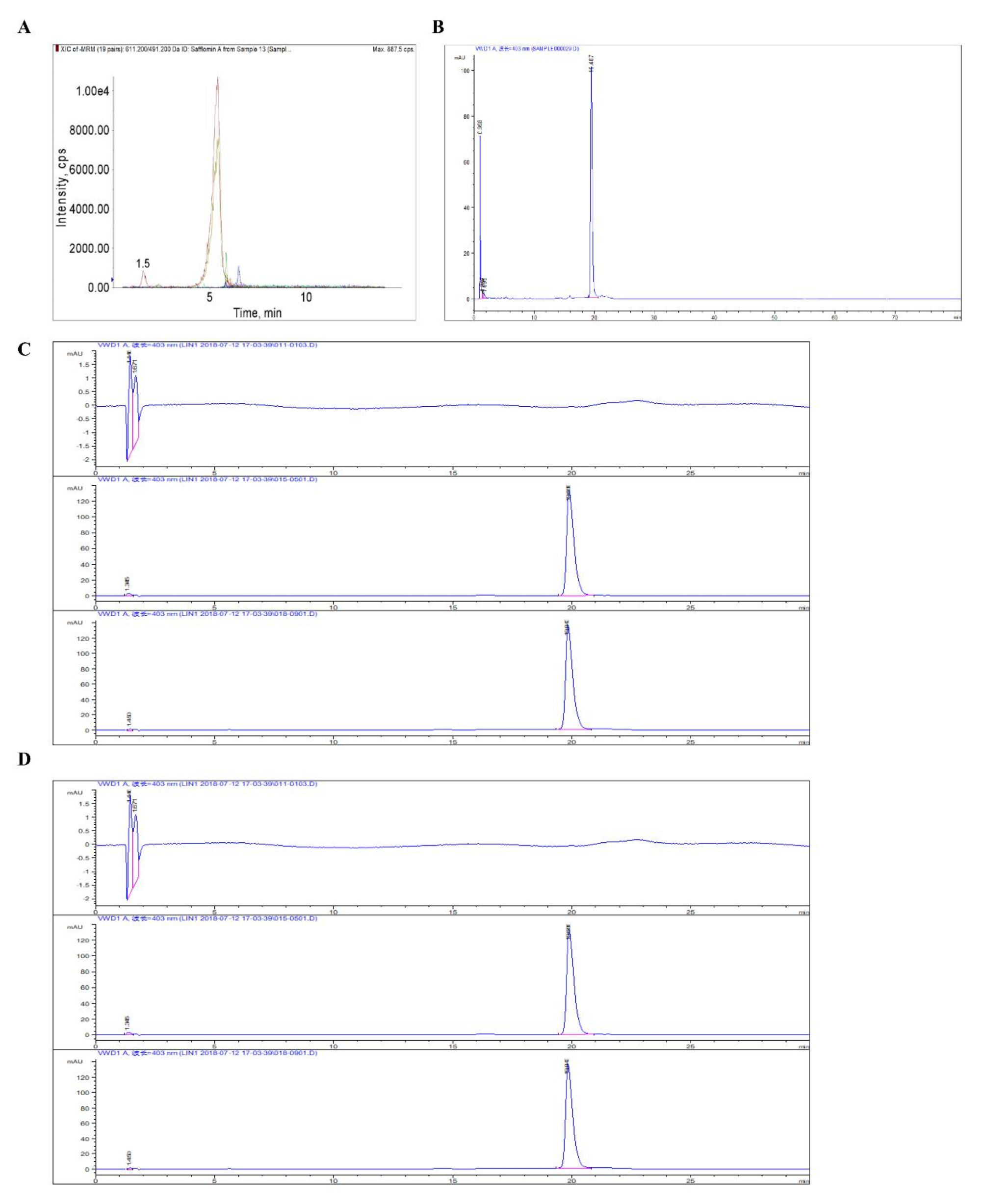
UPLC-Q-TOF/MS analysis of BUDE
BUDE sample precision weighing 10.0 mg, add 70% methanol 1.0 mL, room temperature ultrasonic extraction twice, each time 30 min, 12,000×g centrifugation for 10 min, take the supernatant filtered by 0.2 μm filter membrane, spare. Then weigh the appropriate amount of control product, add methanol to dissolve ultrasonication, 4℃ stored away from light, spare. The analytes were analyzed by ultra-high performance liquid chromatography quadrupole tandem time of flight mass spectrometry (UPLC-Q-TOF/MS) (Waters SYNAPT G2-Si). The chromatographic column was CORTECS®UPLC®T3 (2.1 × 100 mm, 1.6 μm); the mobile phases were 0.1% formic acid in water and pure acetonitrile; the gradient elution: 0–2 min, 5% acetonitrile; 2–32 min, 5-100% acetonitrile; 32–33 min, 100% acetonitrile; 33.5 min, 5% acetonitrile; 33.5–35 min, 5% acetonitrile flow rate 0.3 mL/min, injection volume 2 uL, column temperature 35℃, sample chamber temperature 10℃.
Ion source parameters for mass spectrometry: capillary voltage: positive: 3.0 kV; negative: 2.5 kV; sample cone-well voltage: 40 V; source offset voltage: 80 V; ion source temperature: 120℃; desolventization temperature: positive 500℃, negative 400℃; desolventization flow rate: positive 1000℃, negative 800℃; nebulizing gas pressure: 6.5 Bar.
Mass spectrometry methods: electrospray ESI ion source, positive and negative ion modes were scanned separately, MSE continue full scan mode, scanning time 0.2 s, scanning range m/z 50-1200. Collision energy was used in MSE, the low collision energy was 6 V, and the high collision energy was 15–45 V. Sodium formate was used for the mass spectrometry calibration, and the leucine enkephalins (positive ion mode m/z 556.2771, negative ion mode m/z 554.2615) were used for real-time mass calibration.
Blood pressure detection
An intelligent noninvasive blood pressure monitor (BP-2010AUL) was utilized to take the rats’ blood pressure once a week. The process began by setting the temperature of the blood pressure chamber to 26 ± 1 °C. The rats to be tested were then placed in the chamber and allowed to acclimatize for at least 15 min. Following this, the rats were immobilized in appropriately sized cages equipped with a thermostat, and the temperature was adjusted to 40 °C. Eventually, the rats’ tails were secured through the holes in the cage and inserted into the system’s pulse sensor. After the signal wave had steadied, the device pressurized automatically to gauge and document diastolic blood pressure (DBP), systolic blood pressure (SBP), and mean arterial pressure (MBP).
Detection of serum glycolipid level
After the rats were fasted for 12 h, but still had access to water or alcohol, blood was obtained and placed in a 37℃-water bath for 30 min. After that, it was centrifuged at 3500 r·min− 1 for 10 min to separate the serum. The serum was then tested for fasting blood glucose (FBG), triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-c), and high-density lipoprotein cholesterol (HDL-c) using a reagent kit (Medicalsystem Biotechnology Co., Ltd. China) with a Fully automated biochemistry analyzer (TBA-40FR).
Glucose tolerance test
Rats fasted on food but not water/alcohol for 12 h and then their tail tips were pricked with a blood collection needle. The initial blood droplet was removed using a sterile gauze, and the subsequent droplet was then applied to the test area of the glucose test strip. Subsequently, glucose (2.0 g/kg body mass) was administered by gavage, and the glucose concentration of the rats was measured by a glucometer (Sinocare, China) at 30, 60, and 120 min following the administration of glucose. Moreover, the trapezoidal rule was utilized to calculate the area under the blood glucose curve (AUC) in order to more accurately evaluate glucose tolerance and assess β-cell function.
Enzyme-linked immunosorbent assay (ELISA)
Appropriate amount of serum was taken and routinely thawed to determine the levels of serum Interleukin-6 (IL-6), lipopolysaccharide (LPS) and Fasting Insulin (FINS) by ELISA, and the supernatants of cultured RAW264.7 cells were collected to determine the levels of interleukin-1β (IL-1β) and IL-10 by ELISA. The experimental procedure was performed in strict compliance with the instruction manual of the kit (Jiangsu Meimian Industry Co., Ltd). The insulin resistance index (IRI) and insulin sensitivity index (ISI) were calculated from the corresponding data. IRI = FBG×FINS/22.5 and ISI = ln(1/(FBG×FINS)).
Histological analysis
The pancreas and liver of rats were washed using phosphate-buffered saline (PBS) and then preserved in 4% paraformaldehyde. Afterwards, the tissues were routinely dehydrated, paraffin-embedded, and were cut into 4 μm thick sections. Hematoxylin and eosin staining (H&E) was performed according to standard procedures to visualize morphologic alterations of the liver and pancreatic tissues, which were then photographed under a microscope (MF43-N, China).
Histological evaluations and immunohistochemistry (IHC)
For immunohistochemistry, rat liver and pancreas tissue sections were dewaxed and hydrated, repaired with sodium citrate antigen repair solution, blocked with endogenous peroxidase (ZSGB-BIO, China) according to the instructions, inhibited for ten min using an immunostaining blocking solution. The primary antibody IL-6 (R1412-2, HUABIO) or IL-1β (66737-1-lg, Proteintech) dilution (1:500) were added dropwise and stored in the refrigerator at 4℃ overnight. Subsequently, the corresponding secondary antibody was added dropwise and incubated in an oven at 37 °C for 30 min, followed by DAB color development, hematoxylin staining of the nucleus, and finally dehydrated transparent sealing solution. The expression of liver tissue as well as pancreatic IL-6 and IL-1β were observed under the microscope.
Immunofluorescence (IF)
For pancreatic immunofluorescence, the rat pancreatic tissue sections were deparaffinized, repaired with sodium citrate antigen repair solution, closed with immunostaining blocking solution for 10 min. The sections were then added with primary antibody IRS2 (R382966, Zenbio, 1:200), INS Monoclonal antibody (66198-1-Ig, Proteintech, 1:250) and Glucagon polyclonal antibody (15954-1-AP, Proteintech, 1:500) mixture overnight at 4℃. After washing with PBS three times, rabbit secondary antibody (SA00013-4, Proteintech) was added dropwise at 37℃ for 30 min. For fluorescent double-stained sections, the procedure was repeated with mouse secondary antibody (HA1128, HUABIO) after three rinses with PBS. Finally, antifluorescence quencher containing DAPI was added, a clean coverslip was placed over the antifluorescence quencher, the position of the coverslip was adjusted, and after 10 min, the islet cells were observed and photographed under a microscope (Zeiss SteREO Discovery. V20, Germany).
For RAW264.7 cells immunofluorescence, LPS-induced RAW264.7 cells were washed with PBS at a density of 2×l05 per well in 12-well dishes with cell slides. Next, the cells were immobilized by exposing them to 4% paraformaldehyde for a duration of 20 min at ambient temperature. Following three PBS washes, the cells were permeabilized with membrane-breaking working solution (TritonX-100, 0.1%) for 20 min at ambient temperature. 5% bovine serum albumin (BSA) was blocked at room temperature for 2 h. The cell slides were treated with prepared antibody against NF-κB (ET1603-12, HUABIO) overnight at 4 °C in a humidified box. After being rinsed with PBS, the cells were subjected to treatment with the identifying secondary antibody to the primary antibody and left to stand at room temperature for an hour. Then antifluorescence-quenched seals containing DAPI were added, and the cells were left to incubate at room temperature for 10 min, shielded from light, and stored at 4 °C. All cells were imaged by laser confocal microscopy (Carl Zeiss, Germany).
Liver glycogen content assay
The glycogen content in rat liver was assessed using a commercial kit supplied by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). In summary, the liver samples were thawed from a -80 °C refrigerator, promptly weighed, homogenized with three times the volume of alkaline solution, and then hydrolyzed by heating for 20 min in a boiling water bath. The cooled hydrolysate was diluted with distilled water to achieve the 5% concentration necessary for determining liver glycogen. Following this, the liver glycogen content was measured at 620 nm.
Quantitative real-time polymerase chain reaction PCR (qRT-PCR)
Animal samples consisted of rat liver tissues, collected in appropriate amounts, while cell samples were obtained from RAW264.7 cells after differentiation induction with LPS and drug administration as described above. Both RAW264.7 cells and rat liver tissues have their whole RNA extracted using the Steady Pure Rapid RNA Extraction Kit (Accurate Biology). The RNA concentration of each sample was then determined using an ultra-micro spectrophotometer. Subsequently, single-stranded cDNA was created by reverse transcribing 1 µg of total RNA following the instructions of the MonScript™ RTIII Super Mix with dsDNase (Two-Step) (Monad). The resulting cDNA was then used for quantitative PCR in a real-time fluorescence quantitative PCR instrument (StepOnePlus) with MonAmp™ SYBR ® Green qPCR Mix (High ROX) (Monad). Semi-quantitative analysis was conducted using the ΔΔCt method, and finally the data were normalized using the RNA expression level of β-Actin. The gene sequences of each primer are detailed in the following Table 1.
Table 1 Primer sequence listWestern blotting (WB)
The cells/tissues were lysed in a lysis solution containing protease and phosphatase inhibitors. Subsequently, proteins were separated via SDS-PAGE and then moved onto a polyvinylidene difluoride membrane. The membranes were then incubated in phosphate buffer solution containing 2.0% Tween (TBST) and 20% BSA for 2 h. Following this, the membranes were then treated with IκB (YT2419, Immunoway), p-IκB (YP1372, Immunoway), NF-κB (HA721307, HUABIO), p-NF-κB (db7996, Diagbio), and β-actin (YT0099, Immunoway) for an entire night at 4 °C. The following day, the membranes were washed in TBST and exposed to the appropriate secondary HRP-coupled antibody for 2 h at room temperature. Ultimately, a chemiluminescence detector and an enhanced chemiluminescence solution (ECL) were used to see the protein bands. The enhanced chemiluminescence solution was rinsed off with TBST, then the previously incubated antibody was eluted with stripping buffer (Solarbio, sw3022), rinsed again with TBST, and the primary antibody incubation and subsequent procedure described above was repeated.
Statistics
Statistical analysis was conducted using SPSS 25.0 software. The data were presented as mean ± standard deviation (± s). Group comparisons were performed using one-way ANOVA and t-tests. Statistical significance was denoted by P < 0.05 and P < 0.01.






留言 (0)