Marmosets
Six (six males) common marmosets (Callithrix jacchus; weighing 280–408 g) were obtained from the Osong Medical Innovation Foundation (Chungbuk, Republic of Korea) and housed in marmoset cages [animal number/date of birth/sex (M: male): Vehicle group 1/29Jun20/M; Vehicle group 2/16Feb19/M; Vehicle group 3/21Jun18/M; Cocaine treated group 1/23Dec18/M; Cocaine treated group 2//27Jun18/M; Cocaine treated group 3/17Sep17/M; (45 × 60 × 60 cm)]. The animal room was maintained at a temperature of 27 ± 2 ℃, relative humidity of 40 ± 10% and a 12:12 h light: dark cycle (lighting 07:00–19:00, lighting ≥ 500 Lux) with 10–15 times air exchange. The animals received breeding/maintenance feed (50 g/day, NO. 0630, Altromin, Lage, Germany) prepared for marmosets and sterile tap water ad libitum from an automatic drinker. Cocaine (Cocaine hydrochloride; MacFarlan Smith Ltd., Edinburgh, UK) was administered subcutaneously for 4 weeks at 10 mg/kg. Vehicle group = Control group, Cocaine treated group = Cocaine group.
Mice
Male WT mice (11 weeks old) were purchased from Samtako Bio (Osan, Republic of Korea). Goldenticket (Tmem173 gt) mice are chemically induced mutants of the Tmem173 (Sting) protein, with a missense mutation in exon 6 that changes isoleucine to asparagine at amino acid 199. In homozygous mice, Western blot analysis of bone marrow-derived macrophages showed no detectable protein. However, these mice are viable and fertile, and their peritoneal macrophages do not produce IFN-β in response to c-di-GMP or Listeria monocytogenes infection (Sauer et al. 2011). The C57BL/6 J-Sting1gt/J(STINGgt/gt) mice were provided by Professor Chan Kim (PhD, MD, CHA University School of Medicine, Seongnam, Republic of Korea). All the mice had a C57BL/6 J background. Mice were housed in the facility maintained with a 12–12 h light–dark cycle at 21 ± 2 °C and then transferred to a specific pathogen-free facility. The laboratory maintained high pressure to prevent equipment contamination.
In the first model, WT mice (11 weeks old) were housed at 3–5 per cage. Cocaine was administered at a dose of 20 mg/kg for 10 days intraperitoneally, and saline was administered (intraperitoneally) as a vehicle. A previously published chronic binge-eating ethanol supply protocol was followed to establish an ethanol diet model(Bertola et al. 2013). Briefly, mice were fed a Lieber DeCarli Regular Control Rat Diet (NO. D710027; Dyets, Pennsylvania, USA) for the first five days as an acclimation phase. Control groups were fed a regular control rat diet, and ethanol groups were fed a Lieber DeCarli Ethanol Rat Diet (NO. D710260; Dyets, Pennsylvania, USA) containing 5–6% ethanol (NO. 041237; OCI, SEOUL, Korea) (v/v) were fed each for 10 days. On the morning of day 11, mice were administered a single dose of ethanol (5 g/kg body weight) and sacrificed 9 h later. Vehicle group = Control group, Cocaine treated group = Cocaine group, Ethanol treated group = EtOH group, Cocaine and Ethanol treated group = Cocaine + EtOH group. The second model was also established by additionally using STINGgt/gt mice under the same conditions as the first model. Vehicle group = Control group, Ethanol treated group = EtOH group, Cocaine and Ethanol treated group = EtOH + Cocaine group.
Cell culture
Immortalized mouse Kupffer cells (ImKCs) at the second passage were purchased from Applied Biological Materials Inc. (Richmond, BC, Canada), and cultured in Dulbecco's Modified Eagle's Medium (DMEM) (NO. 10–013-CVRC; Corning, Jiangsu, China) supplemented with 10% fetal bovine serum (FBS) (NO. 12003C; Sigma, Australia). Hep3B cells (in the second passage) were purchased from American Type Culture Collection (ATCC; Manassas, VA) and cultured in DMEM supplemented with 10% FBS. Cells were incubated in an atmosphere of 5% CO2 at 37 °C and treated with cocaine (250 µM) and mito-TEMPO (NO. SML0737; Sigma) (50 µM, pre-treat 1 h) for 24 h to demonstrate cocaine-induced inflammation.
Primary hepatocytes and KC isolation
To isolate primary hepatocytes, C57BL/6 J mice(10 ~ 11 weeks) were anesthetized with Zoletil (30 mg/kg; Virbac, Carros, France), and a catheter (24G) was inserted into the inferior vena cava (IVC). Livers were perfused with 30 mL EGTA solution (NO. 99,590–86-0; Sigma) maintained at 37 °C using a Masterflex L/S easy-load II (Cole-Parmer Instrument Co., IL, USA). The liver was perfused with 75 mL of enzyme buffer containing collagenase type I (650 μg/mL; Worthington Biochemicals, LA, USA) and collagenase P (50 μg/mL; Roche, Mannheim, Germany). Following this, the liver was dissected, chopped, passed through a 100 μm filter, and washed twice with enzyme buffer. Hepatocytes were then isolated and cultured in complete M199 medium (NO. 10–060-CV; Corning, NY, USA).
To isolate KC from WT and STINGgt/gt mice, the digested hepatocyte suspension was centrifuged at 57 x g for 1 min. The supernatant was then collected and centrifuged at 918 x g for 10 min to obtain the cell suspension. The cell suspension was gently covered with 70% Percoll and centrifuged at 2066 x g for 20 min in an off-brake setting. Non-parenchymal cell (NPCs) were collected, and KCs were isolated using MojoSort™ Streptavidin Nanobeads (BioLegend, San Diego, CA, USA). Isolated KCs were resuspended in RPMI 1640 medium (NO. 10–040-CV, Corning).
HA measurement
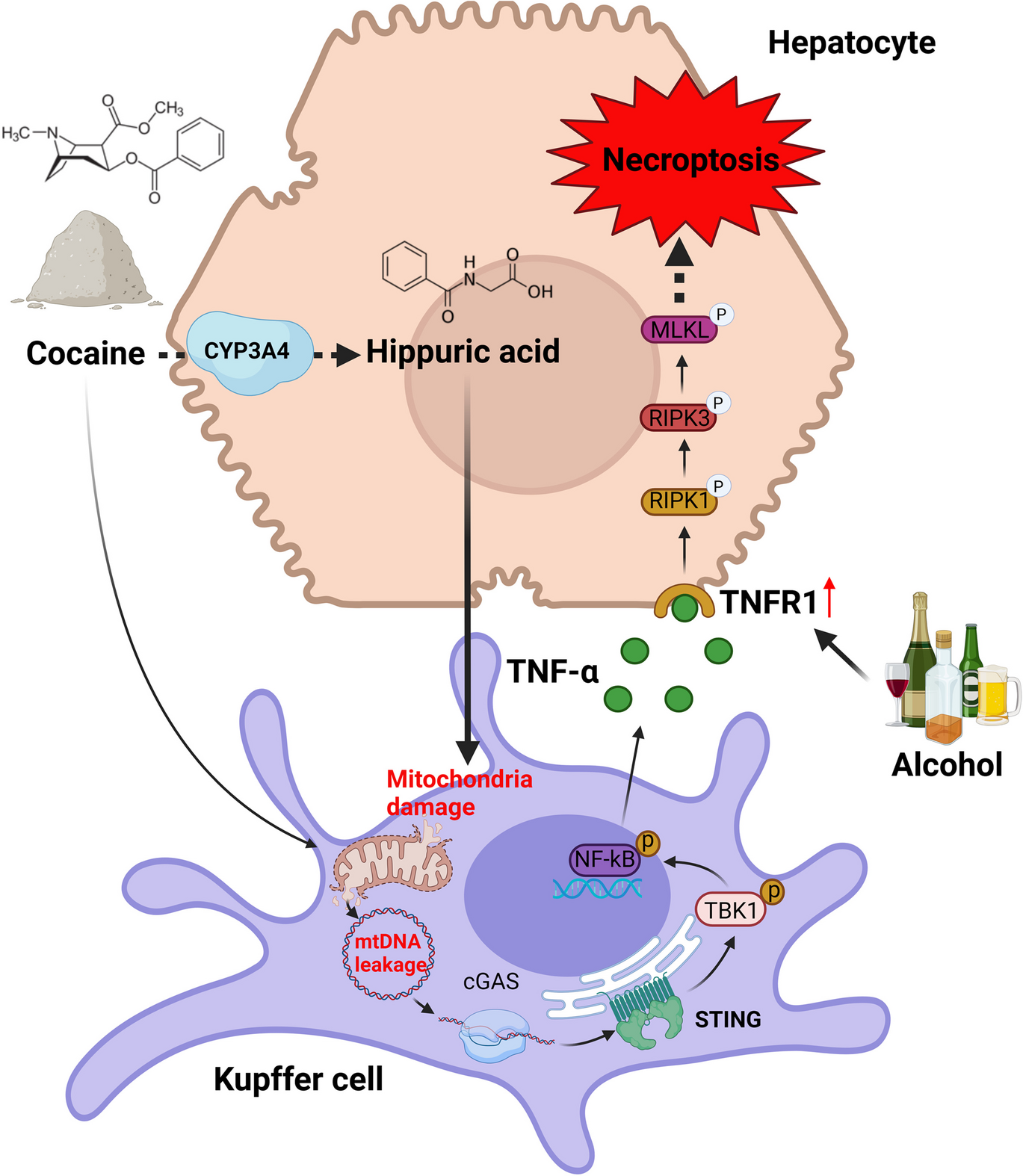
HA in the supernatant was measured using high-performance liquid chromatography (HPLC). The operation was performed on an Alliance e2695 Separations Module (Waters, MA, USA) fitted with a YMC-Pack Pro J'Sphere ODS-H80 column (300 mm × 4.6 mm, 5 μm; YMC, Seongnam, Republic of Korea). The separation was achieved using acetonitrile (A) and water (B) as the mobile phase and the following conditions: 80% A and 20% B for 10 min and 100% B for 11 min; flow rate, 1 mL/min. HA was eluted and detected at around 3.6–3.7 min at 230 nm.
Metabolomics analysis
Marmoset blood samples were collected by performing a cardiac puncture. Blood samples (60 µL) were centrifuged at 13,000 rpm for 10 min, the supernatant (20 µL) was collected, and acetonitrile (100 µL) with internal standard (L-Leusine-5,5,5-d3, 50 µg/mL) was added. The mixture was centrifuged at 13,000 rpm for 10 min; the supernatant was used to analyze metabolites using an Agilent 1290 infinity Ultra High Performance Liquid Chromatography (UHPLC) system (Agilent, Santa Clara, CA, USA) coupled with QTRAP 6500 + (hybrid triple quadrupole linear ion trap mass spectrometer) system (SCIEX, Framingham, MA, USA) and separated using a BEH Amide column (2.1 × 100 mm, 1.7 µm) (Waters, Milford, MA, USA). Mobile phases A (10 mM ammonium acetate and 0.1% acetic acid in water) and B (1 mM ammonium acetate and 0.1% acetic acid in acetonitrile) were used at a flow rate of 400 µL/min. Gradient of mobile phase were used as follows: 15% B and 85% A at 0 min, 17.5% B and 82.5% A at 3 min, 30% B and 70% A at 5.5 min, 46% B and 54% A at 7 min, 54% B and 46% A at 7.5 min, 80% B and 20% A at 9.5 min, and 85% B and 15% A at 10 min. The mass spectrometer was operated in Multiple Reaction Monitoring (MRM) in positive mode. The data were collected and analyzed using MultiQuant 3.0 and MarkerView 1.3 software (SCIEX). Metabolomics Relative change (%) was calculated as follows:
$$\text(\%) =\,(\text - \text)/\text\times 100.$$
p-Toluenesulfonyl chloride colorimetric assay
Mouse blood samples were diluted tenfold with NFW, and 100 μL aliquots were placed into PCR tubes. Each tube received 20 μL of pyridine, followed by 30 μL of a 0.5 M p-TsCl solution (95.33 mg p-TsCl in 1 mL EtOH). The mixture was thoroughly combined until no solids remained visible. 100 μL of each mixture (mouse serum + pyridine) was transferred to a 96-well plate and 0.5 M p-TsCl solution was added, and the absorbance at 570 nm (A570) was measured 2 min later using a microplate reader (Bhattacharyya et al. 2023).
Mitochondrial membrane potential analysis
Mitochondrial membrane potential experiments were performed using JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine iodide) mitochondrial membrane potential assay kit (Cayman Chemical, Ann Arbor, USA). ImKC cells were cultured in the presence of cocaine (250 µM) for 24 h. Afterward, the cells were treated with JC-1 fluorescent dye at 37 °C for 20 min and washed twice with Dulbecco's phosphate buffered saline (DPBS) buffer (100 μL/mL). Fluorescence was measured using a Microplate Reader (Tecan, Männedorf, Switzerland) (red: excitation 550 nm, emission 600 nm; green: excitation 485 nm, emission 535 nm). Mitochondrial depolarization was assessed by measuring the red-to-green fluorescence ratio.
Flow cytometry
ImKC cells were treated with cocaine (250 µM) for 24 h to induce mitochondrial ROS (mtROS) production. Subsequently, the cells were stained with 5 μM MitoSox™ Red mitochondrial peroxide indicator (Invitrogen, Waltham, USA) according to the manufacturer's recommendations. After treatment, the fluorescence intensity of MitoSox™ Red was measured at 580° using a FACS Calibur flow cytometer (Guava® easyCyte™ HT System, Luminex, Austin, USA). Data were analyzed using the Guavasoft software (Luminex, Austin, USA).
Cytotoxicity analysis
Isolated primary hepatocytes were cultured in collagen-coated 12-well plates (2 × 105 cells/well) in filtered M199 medium supplemented with 10% FBS and 1% antibiotic and antifungal agents (Gibco, NY, USA). After 3 h, the medium was replaced with KC-derived conditioned medium (CM) without FBS. After incubation for 24 h, the medium was collected for further analysis. Cytotoxicity was measured using a Quanti-LDH™ Cytotoxicity assay kit (Biomax, Seoul, Republic of Korea).
mtDNA copy number analysis
DNA was isolated from ImKC cells using the MagMAX Cell-Free DNA (cfDNA) Isolation kit (Invitrogen). The DNA was subjected to real-time polymerase chain reaction (PCR) in a CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) using SYBR qPCR master mix (TB Green, TAKARA Bio, Kusatsu, Japan) to determine the mtDNA/nuclear DNA ratio to determine the number of mtDNA copies. The primers for real-time PCR are listed in Table S2.
Small interfering RNA (siRNA) transfection
Primary hepatocytes were cultured to 30–50% confluence in 6-well culture dishes. Commercially available mouse scrambled siRNA (IDT, Coralville, IA, USA) and TNFR1 siRNA (IDT, Coralville, IA, USA) were transfected into the cells using Lipofectamine® 3000 (Thermo Fisher Scientific Inc., Waltham, CA, USA) according to the manufacturer's instructions.
Enzyme-linked immunoassay (ELISA)
Protein levels of TNF-α and IL-1β after stimulation with cocaine in ImKC cells for 0, 3, 6, 12, and 24 h were determined using ELISA. The ELISA was performed using Mouse TNF-alpha DuoSet and Mouse IL-1 beta/IL-1F2 DuoSet (DY401, R&D Systems, Minneapolis, Minnesota, USA) kits according to the manufacturer's instructions. The absorbance was measured at 450 and 540 nm using a microplate reader (VersaMax Absorbance Microplate Reader, Molecular Devices). Absorbance was calculated by subtracting the absorbance at 540 nm from that at 450 nm. A four-parameter regression analysis was used to create a standard curve.
Quantitative real-time PCR analysis
Cells and liver tissues were homogenized using RiboEx (NO. 301–001). RNA was isolated from the cells and liver tissues using a Hybrid-R RNA extraction kit (Gene All Biotechnology, Seoul, Republic of Korea). Reverse transcription was performed using RNase-free DNase I (Promega, Madison, WI, USA) and a Capacity cDNA Reverse Transcription Kit (Applied Biosystems, FC, USA) according to the manufacturer's instructions. cDNA was subjected to real-time PCR using a CFX Connect Real-Time PCR Detection System (Bio-Rad) and SYBR qPCR master mix (TB Green)—the primers used are listed in Table S2. Quantification was performed by normalizing the expression of the target gene to that of glyceraldehyde 3-phosphate dehydrogenase (Gapdh; internal control).
Western blot analysis
ImKC cells, primary hepatocytes, and liver tissues were homogenized in RIPA buffer. The lysates were centrifuged at 13,000 rpm for 15 min at 4 °C. Protein concentration in the supernatant was measured using a BCA Protein Assay Kit (Thermo Fisher Scientific Inc.). Equal protein amounts were separated by 10% SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 5% skim milk for 1 h at 21–23 °C and with 5% BSA, then incubated overnight at 4 °C. The membrane was then incubated with primary antibodies (1:1000) overnight at 4 °C, followed by secondary antibodies conjugated with horseradish peroxidase at room temperature for 1 h. All antibodies were diluted in Tris-buffered saline/Tween with 2% BSA.
KC conditioned medium (CM) delivery experiment
KCs (2.5 × 105 per well) were incubated in 6-well plates with or without cocaine (250 µM) for 24 h. The KCs supernatant was harvested with pipet, and the remaining cells were pelleted at 600 × g for 5 min, then transferred to hepatocytes (2.5 × 105) in a 6-well plate and incubated at 37 °C. for 24 h.
Biochemical analysis
Mouse blood samples were collected via cardiac puncture and centrifuged at 13,000 rpm for 15 min to obtain serum. AST and ALT levels in the serum were analyzed using a biochemical analyzer (KP&T, Osong, Republic of Korea), while AMY, ALP, and LIPA levels were measured using an Exdia PT10V (Precision Biosensor, Daejeon, Republic of Korea). Marmoset blood samples were taken immediately before the first and after the last cocaine self-administration session, as well as during two consecutive instances of stereotyped behaviors. Blood was collected in EDTA- or heparin-treated tubes (approximately 24 h after cocaine administration), centrifuged at 2,000 × g for 15 min at 4 °C, and the plasma was transferred to microtubes and stored at − 80 °C until analysis.
Histopathological analysis of liver
To collect livers, mice were anesthetized with Zoletil (30 mg/kg). The extracted liver tissues were cleaned, fixed in formalin, and embedded in paraffin. Tissue blocks were cut into 5 μm-thick sections and stained with hematoxylin and eosin (H&E) (NO. 3,801,698, Leica Biosystems). To confirm hepatocyte necrosis, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using ApopTag® Plus Peroxidase in Situ Apoptosis Kit (NO. S7101, Millipore, Burlington, USA).
Statistical analysis
All data are expressed as the mean ± standard error of the mean (SEM). Statistical analysis by t-test was performed using GraphPad Prism 8 software (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was set at p < 0.05. made available on request.



留言 (0)