
記住我
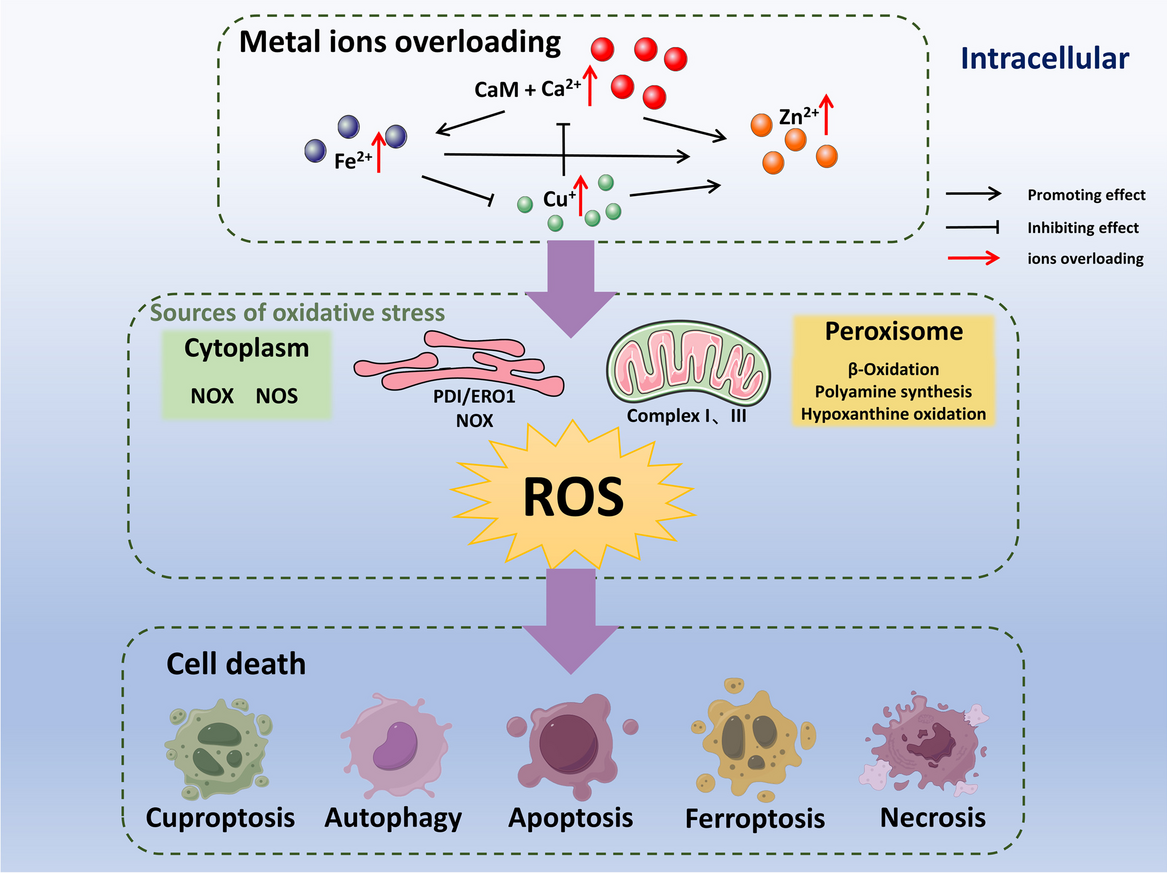
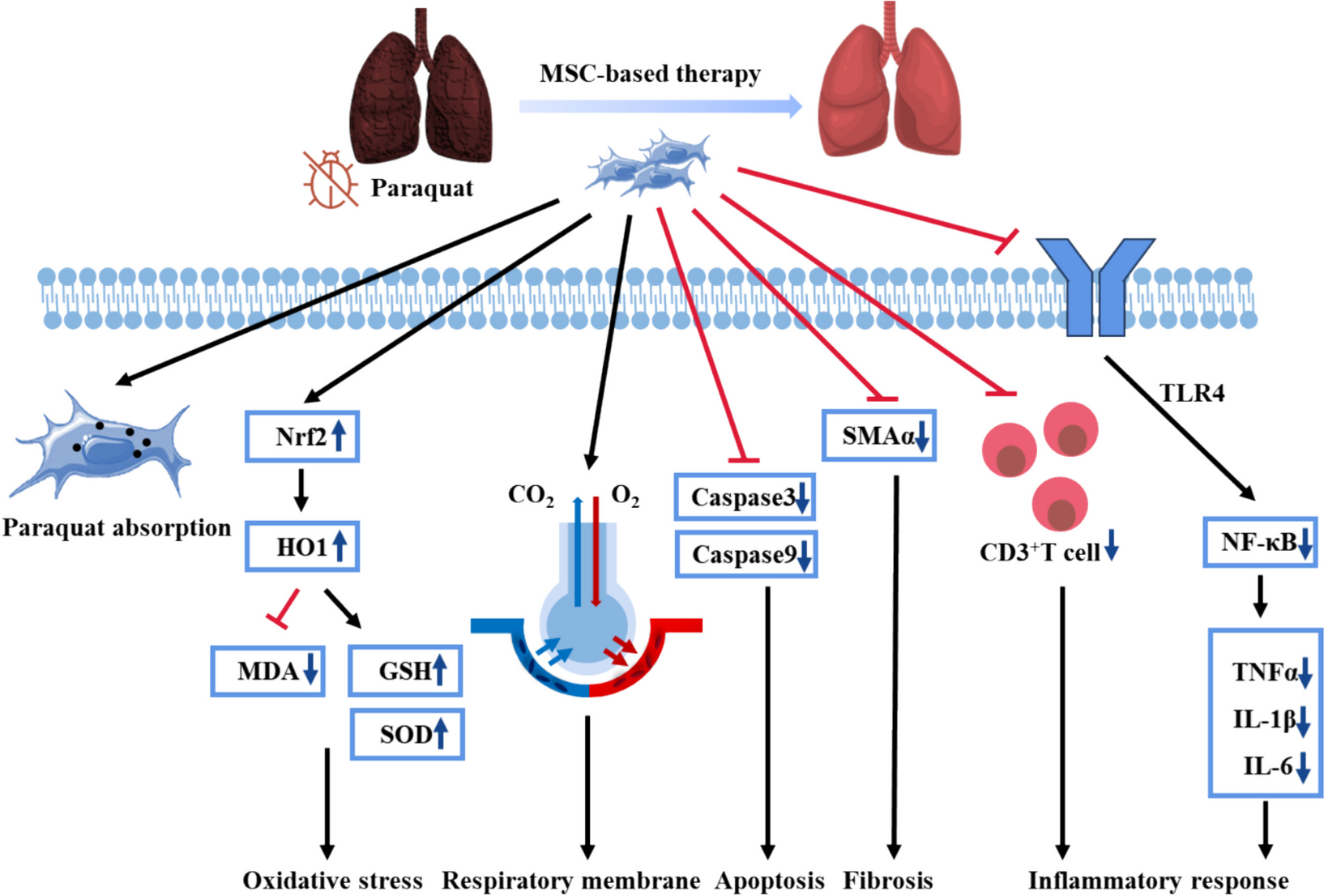
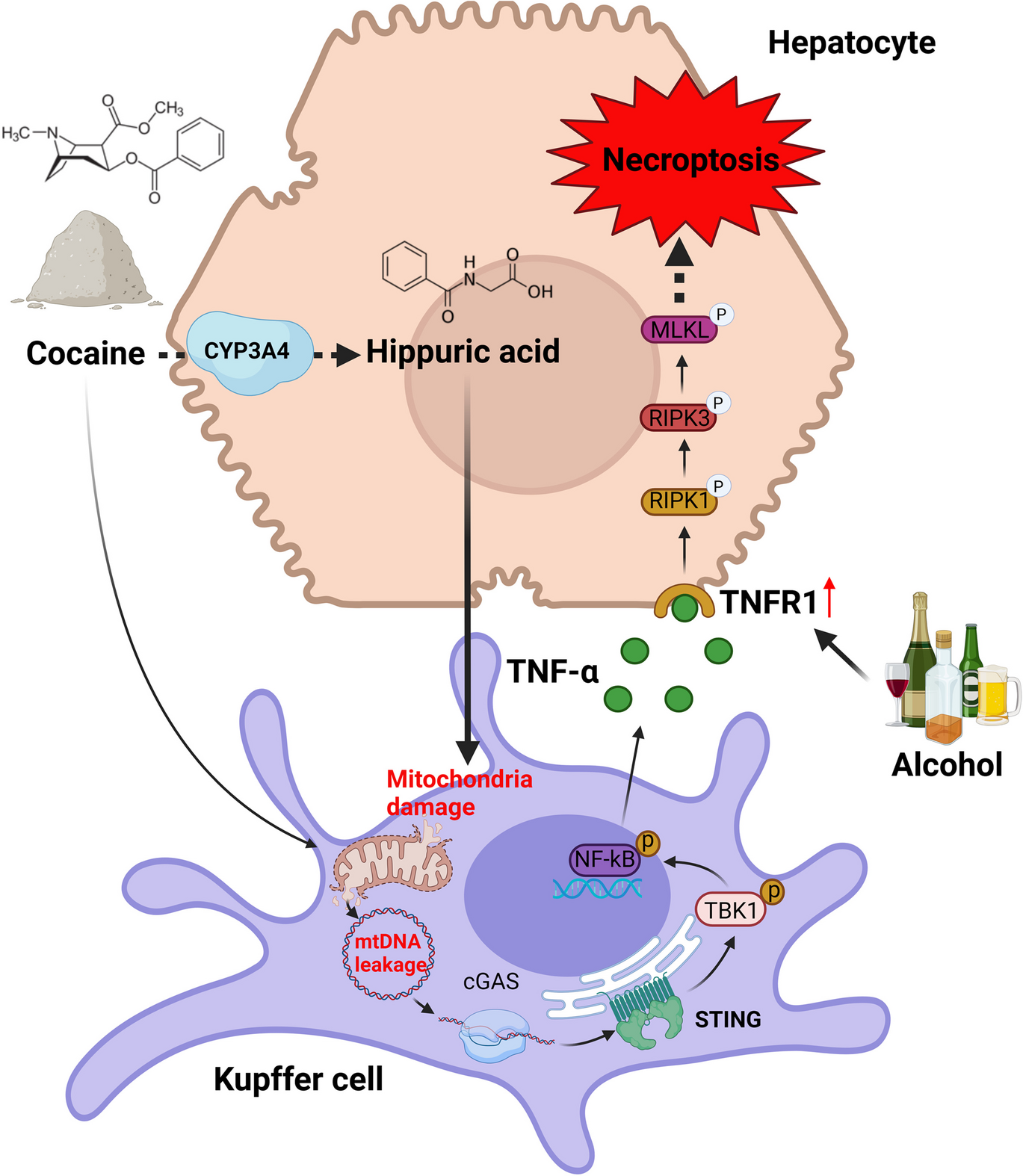
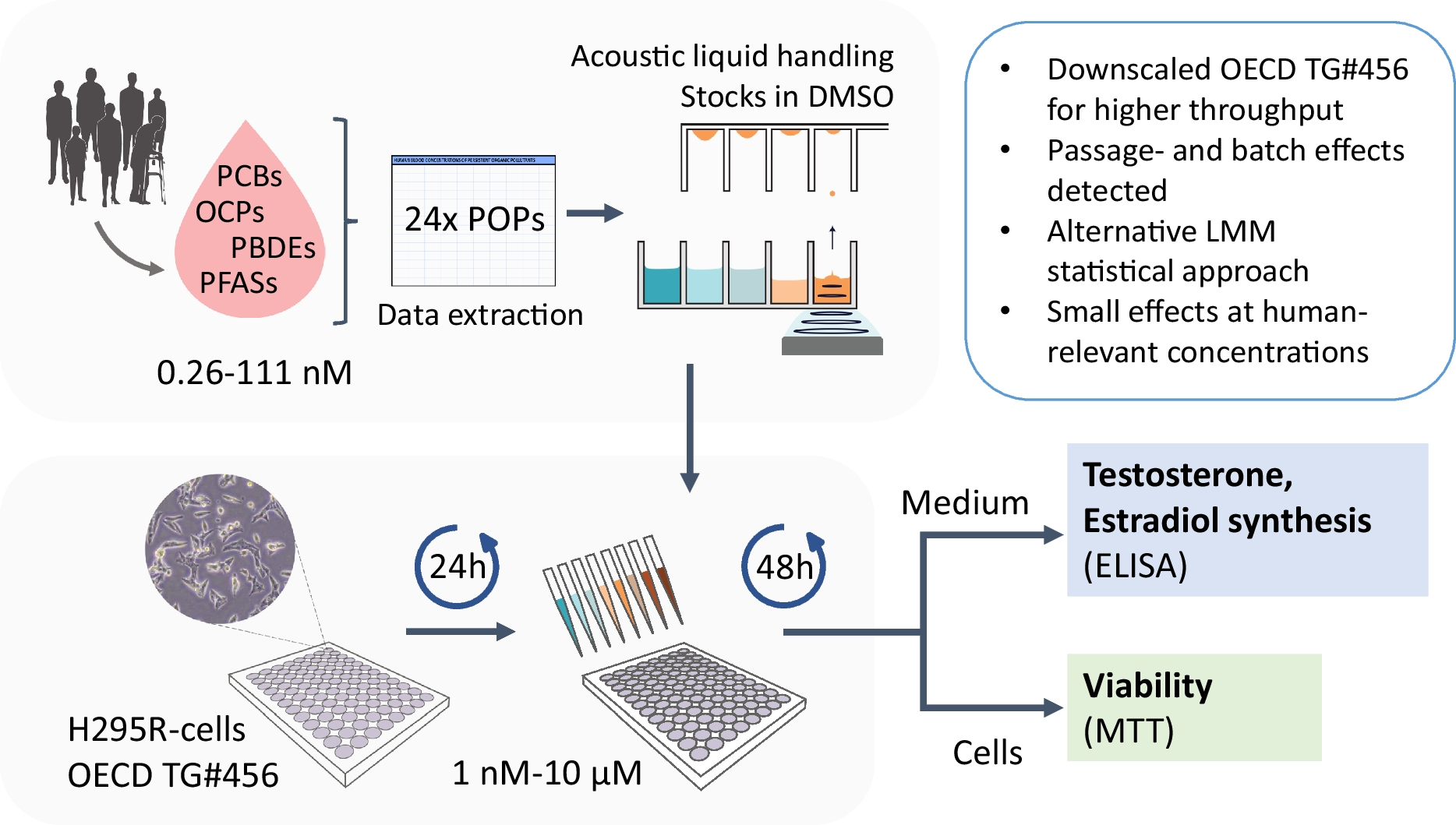


This study was carried out with the approval and oversight of the Institutional Review Board of Rongchang District People’s Hospital. The animal experimental procedures were approved by the Animal Ethics Committee. The clinical aspects of this research involving human participants were reviewed and approved by the Clinical Ethics Committee with the ethical. Informed consent was obtained from all subjects.
Lactobacillus Plantarum was cultivated, and its outer membrane vesicles were extracted, isolated, and identifiedThe strain of L. plantarum (Bio-67374) we obtained was derived from plants and sourced from the Chinese Microbial Strain Resource Database. A 1% inoculum was added to 10 mL of sterilized MRS broth (CM0359B, Thermo Fisher Scientific, Waltham, MA, USA), followed by incubation at 37℃ for 24 h. Subsequently, the bacterial suspension was mixed with 50% glycerol at a 1:1 ratio and stored frozen at -80 °C. To assess growth and pH variations, 5% Lactobacillus plantarum was inoculated into MRS broth and cultured at 37 °C for 48 h, with bacterial counts measured at 0, 8, 24, and 48 h. Bacterial counts were measured at 0, 8, 24, and 48 h. The formula for calculating the bacterial colony count is obtained by dividing the colony count by the dilution volume and then multiplying the result by the dilution factor. The samples are collected at 0, 2, 8, 16, 24, and 48 h. Subsequently, the pH of the culture medium is measured using a pH meter.
Extraction and separation of LEVs: L. plantarum was cultured until it reached the stationary phase with colony forming units (CFU) of ≥ 2.7 × 109 CFU/mL L. plantarum. The culture was then subjected to consecutive centrifugation at 1800 × g at 4 °C for 20 min and 10,000 × g at 4 °C for 20 min to collect the bacterial pellet and supernatant, respectively. Filtering was conducted using a cellulose acetate membrane (HAWG0470, Millipore, Merck, UK) with a pore size of 0.45 μm. Concentration was accomplished using a Centricon Plus-70 membrane Ultracel-PL (UFC710008, Millipore, Merck, UK) with a molecular weight cut-off of 100 kDa. The concentrated supernatant should be centrifuged at 4 °C and 260,000 × g for one hour. After that, the pellet should be washed twice with phosphate-buffered saline (PBS) or 0.1 M salt-free phosphate buffer (PB). The EV-containing pellet should be resuspended in PBS and stored at -20 °C for further experiments.
To identify LEVs, they are fixed with 1% glutaraldehyde, placed on grids coated with formic acid ester, and restained with 30 μL of 1% phosphotungstic acid solution (pH 6.8) for 5 min at room temperature. Dry the sample using an incandescent lamp and observe it under a JEM-2000EX transmission electron microscope (JEOL, Japan), capturing images at the appropriate magnification. The Zetasizer Nano ZS90 (Malvern, UK) was utilized for quantifying and analyzing the size distribution of extracellular vesicles (EVs) (Kim et al. 2020; Bajic et al. 2020; Lee et al. 2021; Li et al. 2017; Yang et al. 2022).
Cell cultureWe utilized the following human colon cancer cell lines: CaCo-2 (CC-Y1089), SW480 (CC-Y1500), and HCT-116 (CC-Y1194). Additionally, we employed the human normal colon epithelial cell line NCM460 (CC-Y1550) (1) and the human embryonic kidney cell line 293 T (CC-Y1010) (1). All cell lines were acquired from Enzyme Research Biotechnology in Shanghai, China. The CaCo-2 and 293 T cells were cultured in high-glucose DMEM medium (11,965,084, Thermo Fisher Scientific, USA) supplemented with 20% or 10% FBS (26,140,079, Thermo Fisher Scientific, USA) and 1% antibiotic–antimycotic solution (100 U/mL penicillin and 100 μg/mL streptomycin) respectively. The SW480, HCT-116, and NCM460 cells were cultured in Leibovitz's L-15 Medium (11,415,064, Thermo Fisher Scientific, USA), McCoy's 5A Medium (16,600,082, Thermo Fisher Scientific, USA), and RPMI-1640 (11,875,101, Thermo Fisher Scientific, USA) respectively, supplemented with 10% FBS and 1% antibiotic–antimycotic solution. All cell cultures were conducted in a temperature-controlled incubator (model BB15, Thermo Fisher Scientific, USA) at 37 ℃ with 5% CO2 (Li et al. 2017).
Cellular uptake of Lactobacillus plantarum-derived extracellular vesiclesThe LEVs were labeled with PKH26 green fluorescent dye (MINI26, Sigma-Aldrich, USA) and then incubated at room temperature for 5 min with a concentration of 10 μg/ml of the labeled LEVs (Xian et al. 2019). The cells were suspended in a standard culture medium and incubated with Caco-2 cells at 37 °C for 24 h. Then, wash the cells twice with PBS. DAPI is used to label cell nuclei. Stained cells were observed using the IX53 fluorescence microscope (CLS; LSM 510 META, Carl Zeiss AG) (Cheng et al. 2018; Wei et al. 2020).
RNA extraction and sequencingLactobacillus plantarum cells were collected by centrifugation at 12,000 g for 5 min. Then, the cells were suspended in a DMEM high glucose medium supplemented with 1% non-essential amino acids until an optical density of 0.9 at 600 nm was reached. Caco-2 cells were treated with a cell suspension containing Lactobacillus plantarum cells or a control medium for 10 h. Subsequently, two groups of cell samples were collected, each containing four samples, and total RNA was isolated using Trizol Reagent (15,596,026, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
The concentration and purity of the RNA samples were measured using a Nanodrop 2000 spectrophotometer (1011U, Nanodrop, Thermo Fisher Scientific, Waltham, MA, USA). Subsequent experiments utilize total RNA samples that meet the following criteria: RNA Integrity Number (RIN) ≥ 7.0 and a 28S:18S ratio ≥ 1.5, determined through denatured agarose gel electrophoresis and Bioanalyzer 2100 analysis for assessing RNA integrity.
CapitalBio Technology, located in Beijing, China, prepared and sequenced the sequencing library. Each sample requires a total of 5 μg of RNA. To eliminate ribosomal RNA (rRNA) from total RNA, the Ribo-Zero™ Magnetic Kit (MRZE706, Epicentre Technologies, Madison, Wisconsin, USA) is employed. The NEB Next Ultra RNA Library Prep Kit (#E7775, NEB, USA) is utilized for Illumina sequencing and library construction. Subsequently, the RNA fragment is fragmented into approximately 300 base pair (bp) fragments using NEB Next First Strand Synthesis Reaction Buffer (5 ×). The first strand of cDNA is synthesized using reverse transcriptase primers and random primers, while the synthesis of the second strand of cDNA takes place in the reaction buffer of dUTP Mix (10 ×) for the second strand synthesis. The repair of cDNA fragment ends involves the addition of a polyA tail and the ligation of sequencing adaptors.
Following the ligation of Illumina sequencing adapters, the second strand of cDNA was digested using USER Enzyme (#M5508, NEB, USA) to construct a strand-specific library. The library DNA should be amplified, followed by purification and enrichment through PCR. Subsequently, the library was analyzed using an Agilent 2100 instrument and quantified utilizing the KAPA Library Quantification Kit (KK4844, KAPA Biosystems). Finally, we conducted paired-end sequencing using the NextSeq CN500 (Illumina) sequencer (Regan et al. 2021; Bao et al. 2023).
Quality control of the sequencing data and alignment to the reference genome should be performedFastQC software version 0.11.8 assessed the quality of paired-end reads in the raw sequencing data. The raw data was preprocessed using version 1.18 of the Cutadapt software to eliminate Illumina sequencing adapters and poly(A) tail sequences. We used a Perl script to discard reads with an N content of over 5%. Reads with a base quality of over 20 were extracted, amounting to 70% of the total, using the FASTX Toolkit version 0.0.13 software. Repair the paired-end sequences using BBMap software. The filtered high-quality read fragments were finally aligned to the human reference genome using hisat2 software (version 0.7.12) (Bao et al. 2023).
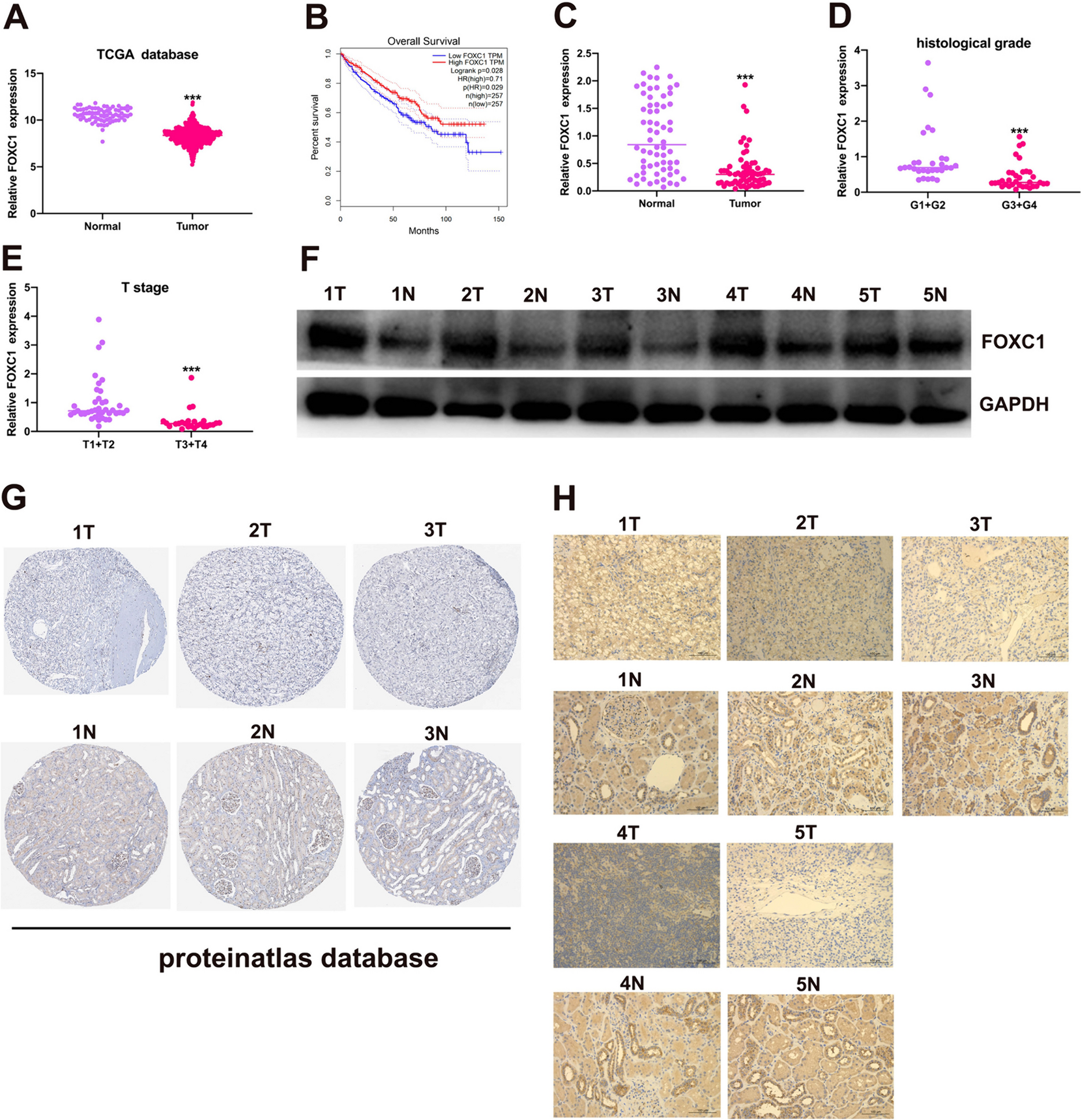
Bioinformatics analysisTranscriptome sequencing data for colon cancer (TCGA-COAD) were obtained from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/), comprising 483 colon cancer tissue samples and 41 adjacent normal tissue samples. The mRNA expression levels were analyzed using the "edgeR" package in R language (version 4.2.1) based on read counts. Differential expression analysis was performed between Caco-2 cell samples and TCGA-COAD dataset samples, applying the criteria |log2FC|> 0 and P.value < 0.05 for selecting differentially expressed genes.
Colon cancer-related genes were retrieved from disease-related search databases, phenolyzer (https://phenolyzer.wglab.org/) and CTD (https://ctdbase.org/), with filtering criteria set at Score > 0.0015 and Inference Score > 25, respectively. The R packages ggplot2 (version 3.3.6) and VennDiagram (version 1.7.3) were employed to generate and visualize a Venn diagram to obtain the intersection between differential genes and disease-related genes. The STRING database (https://string-db.org/) analyzes protein–protein interactions encoded by genes. For visualizing the protein–protein interaction networks, Cytoscape 3.6.1 software is employed.
The CytoHubba plugin, based on Cytoscape 3.6.1 software, is also utilized to further filter core genes based on MCC values. The dynamic network Venn diagram could be created using the tools available on the Kidio bioinformatics cloud platform. We performed KEGG enrichment analysis on the differentially expressed genes obtained from transcriptome sequencing of Caco-2 cells using the KOBAS database. Genes with a P-value < 0.05 were considered statistically (Liu et al. 2018; Bu et al. 2021).
Plasmid transfection and cell experiment groupingColorectal cancer cells in a healthy state were collected and digested using pancreatic enzymes. The cells were then seeded in a 24-well plate at a density of 8 × 103 cells per well and cultured until they formed a monolayer. Subsequently, the culture medium should be removed, and the cells should be transfected using the Lipofectamine 3000 protocol (L3000150, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Following transfection, the cells were cultured at 37 °C with 5% CO2 for 6 to 8 h. After replacing the complete medium, the cells were further cultured for 48 h to allow extraction of RNA and protein for subsequent experiments.
The specific groups are as follows:
(1).The oe-NC group and the oe-SIRT5 group;
(2).The groups included the PBS group, LEVs group, LEVs + oe-NC group, and LEVs + oe-SIRT5 group. The LEVs were administered at a concentration of 1 μg and allowed to act for 24 h.
(3).sh-NC group, sh-SIRT5#1 group, and sh-SIRT5#2 group;
(4).sh-NC group, sh-p53#1 group, and sh-p53#2 group;
(5).control group (PBS + sh-NC), LEVs + sh-NC group, LEVs + sh-p53 group.
Following a 48-h transfection period of colorectal cancer cells with plasmids, certain groups will receive a treatment of 1 μg of LEVs for 24 h. The optimal concentration for each plasmid should be determined by consulting the instruction manual and then adjusted based on the specific circumstances. The overexpression plasmid for SIRT5 (oe-SIRT5) and its control plasmid (oe-NC) was obtained from RayBiotech. The plasmid vector pEXP-RB-Mam (R11091.1, RayBiotech, Guangzhou, China) was used for construction. The following content contains the target sequences for sh-SIRT5#1 (TRCN0000018546) [CGTCCACACGAAACCAGATTT], sh-SIRT5#2 (TRCN0000232661) [AGAATTACAAGAGTCCAATTT], sh-p53#1 (TRCN0000003754) [TCAGACCTATGGAAACTACTT], sh-p53#2 (TRCN0000003753) [CGGCGCACAGAGGAAGAGAAT], and sh-NC [CTCGCTTGGGCGAGAGTAA] (Jin et al. 2017). Purchased from Sigma-Aldrich (USA) (Yang et al. 2020; Ye et al. 2020).
EdU stainingColorectal cancer cells exhibiting favorable growth conditions were cultured in a 12-well plate and transfected. After 48 h, perform EdU detection to analyze cell proliferation. The cells were incubated with the EdU reaction system (C0071S, BeyoClick™ EdU-488 Cell Proliferation Assay Kit, Beyotime, Shanghai, China) solution for 2 h. After that, the cells were fixed with 4% paraformaldehyde and washed three times with PBS. Subsequently, a single wash with 0.5% TritonX-100 was performed. Then, add DAPI solution to stain the cells. After three washes with PBS, images were captured from the fluorescence microscope for the subsequent calculation of the proliferation rate (Yao et al. 2022).
Clone formation experimentColon cancer cells were collected from various groups and re-suspended following digestion with 0.25% trypsin. Inoculate each group of cells, consisting of 200 cells per well, into 10 mL of culture medium. Gently shake the mixture to ensure an even distribution of cells. In the transfection experiment, the fresh culture medium was replaced every two days after transfection, and this replacement occurred for 12 h. Re-transfect cells on day 6. The cells were fixed with a solution of more than 4% paraformaldehyde (P1110, Solarbio, Beijing, China) for 6 days and subsequently stained with a 0.1% crystal violet staining solution (G1063, Solarbio, Beijing, China) for 15 min. Cell colonies containing more than 50 cells should be counted utilizing a stereo microscope (Chen et al. 2020; Zhou and Zhao 2019; Xiang et al. 2019). The experiment is repeated three times.
Flow cytometryColorectal cancer cells were cultured and transfected with corresponding plasmids for 48 h. Subsequently, the cells were transferred to collection tubes, fixed with pre-chilled 75% ethanol at 4 °C overnight. The cells were then washed thrice with ice-cold PBS and resuspended in 100 μL PBS. Following this, 0.5 mL of PI/RNase staining buffer (550,825, BD Biosciences, USA) was added, and the cells were incubated in the dark at room temperature for 15 min. Flow cytometry was performed within 24 h for PI staining. Finally, cell cycle distribution was determined using a flow cytometer (Guava® easyCyte™ 6-2L Base System, 0500–5007, Luminex, https://www.luminexcorp.com/) (Yao et al. 2022).
Lactic acid production and glucose intakeThe lactate production in tissues and cells was measured using the lactate assay kit (MAK064, Sigma-Aldrich, USA). Mix the cellular or tissue samples with a lactic acid analysis buffer at a four times greater volume for dissolution. Centrifuge the mixture at 13,000 g for 10 min. Remove the residue and eliminate LDH using a 10 KDa ultrafiltration centrifuge tube. Subsequently, transfer the resulting supernatant (50 μL) to a 96-well plate. Add 50 μL of lactic acid analysis buffer to each well. Thoroughly mix the contents using a pipette. Next, incubate the mixture at room temperature in a dark environment for 30 min. Finally, use a fluorescence plate reader with Ex/Em = 540/590 nm to monitor the changes in fluorescence intensity. Glucose uptake in tissues and cells was assessed using a fluorescent assay kit (MAK084, Sigma-Aldrich, USA) to determine glucose levels. Colon mucosal cells were isolated from clean-treated mouse intestines and cultured in a glucose-free RPMI medium. Subsequently, the cells were incubated for 60 min under either 1.0 mM or 20 mM glucose conditions to measure glucose uptake. Colon cancer cells were cultured in fresh low-glucose DMEM medium supplemented with 200 μM 2-NBDG. The cells were then incubated for 10 min under either 20 mM glucose or glucose-free conditions to measure glucose uptake (Fan et al. 2018; Lin et al. 2010).
The Co-IP experiment detects protein–protein interactionsTransfect Flag-SIRT5 (plasmid 13,816, addgene, USA) or HA-p53 (plasmid D3033, Beyotime, Shanghai, China) plasmids into 293 T cells to detect exogenous proteins using co-immunoprecipitation (co-IP) assays. Additionally, conduct co-IP experiments to detect endogenous proteins in Caco-2 and SW480 cells.
Initially, the cells were lysed using IP lysis buffer (P0013, BiyunTian, Shanghai, China) enriched with protease and phosphatase inhibitors. The cell lysate was centrifuged at 12,000 g for 20 min at 4 °C. Next, the 293 T cell lysates, which contained 200 μg of protein, were treated with either an anti-Flag antibody (mouse; 1:50, F3165, Sigma-Aldrich, USA) or anti-HA antibody (mouse; 1:50, H9658, Sigma-Aldrich, USA) at 4℃ for 4 h. Either an anti-p53 antibody (mouse; 1:50, 60,283–2-Ig, Proteintech, Wuhan Sanying, China) or IgG (1:50, #3900, Cell Signaling, USA) was added to the lysates of Caco-2 and SW480 cells and incubated overnight at 4 °C. Next, protein A/G Sepharose beads (sc-2003, Santa Cruz, USA) were used to capture the antibody-protein complexes. After thoroughly washing, the complex was boiled in 1 × SDS sample buffer and subjected to Western blot analysis.
The antibodies used for Western blot analysis included rabbit anti-Flag antibody (1:1000, F7425, Sigma-Aldrich, USA), rabbit anti-HA antibody (1:1000, #3724, Cell Signaling, USA), rabbit anti-SIRT5 antibody (1:2000, 15,122–1-AP, Proteintech, Wuhan Sanying, China), and rabbit anti-p53 antibody (1:5000, 10,442–1-AP, Proteintech, Wuhan Sanying, China) (Zhang et al. 2019a, 2019b).
In vitro succinylation experimentTransfect 293 T cells with HA-p53 plasmid. The reaction system comprises an amber acylation buffer containing 20 mM HEPES at pH 8.0, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 0.1 mg/mL bovine serum albumin. Different concentrations (0.1, 0.2, 0.4 mM) of amber acyl-CoA (S1129, Sigma-Aldrich, USA) were used. Incubate the reaction mixture at 30 °C for 15 min. The reaction was stopped by adding a loading buffer, and SDS-PAGE was performed. The succinylated p53 protein and other proteins were examined using co-IP and Western blot analysis. For this purpose, a rabbit pan-Succinyl-K antibody (#PTM-401, 1:1000 dilution, PTM Bio, Hangzhou, Zhejiang) was employed (Liu et al. 2022; Yang et al. 2018).
In vitro desuccinylation experimentThe highly succinylated HA-p53 protein, purified from 293 T cells, was incubated with purified SIRT5 protein (ab101134, Abcam, UK) or Flag-SIRT5 (wild-type or enzymatically deficient mutant H158Y) for 1 h at 30 °C, with or without 1 mM NAD + . The incubation was carried out in succinate dehydrogenase buffer. The composition of the Amber enzyme buffer is as follows: 50 mM Tris–HCl (pH 8.0), 100 mM NaCl, 8 mM MgCl2, 20% glycerol, 1 mM DTT, 1 mM PMSF, and 0.1 mg/mL BSA. After adding the loading buffer, the reaction should be stopped, and then the separation could be carried out using SDS-PAGE. Analyze proteins through co-IP and Western blot. The deacetylation status of p53 was examined in different treatment groups, such as Caco-2 and SW480 cells, mouse colon polyp tissues, and colon cancer patient tissues, using co-immunoprecipitation (co-IP) and Western blot techniques (Liu et al. 2022; Yang et al. 2018).
RT-qPCRTotal RNA was extracted from colon cancer cells using Trizol reagent, and the concentration and purity of the extracted RNA were measured using a Nanodrop 2000 microvolume UV–Vis spectrophotometer. The RNA was reverse transcribed to synthesize cDNA using the protocol specified in the PrimeScript RT reagent Kit (RR047A, Takara, Japan). The conditions for reverse transcription are as follows: incubate at 42℃ for 30 to 50 min, followed by heating at 85℃ for 5 s. qRT-PCR detection was done using the Fast SYBR Green PCR kit (RR820A, Takara, Japan) and the ABI PRISM 7300 RT-PCR system (Applied Biosystems). The reaction conditions were as follows: pre-denaturation at 95 °C for 5 min, denaturation at 95 °C for 30 s, annealing at 57 °C for 30 s, and extension at 72 °C for 30 s, with a total of 40 cycles. Each hole is set with 3 repetitions. β-Actin is utilized as an internal reference, and the 2−ΔΔCt method is employed to assess the relative expression level of the gene. ΔΔCt is calculated as the difference between the average Ct value of the target gene in the experimental group and the average Ct value of the housekeeping gene in the experimental group, subtracted from the difference between the average Ct value of the target gene in the control group and the average Ct value of the housekeeping gene in the control group. The experiment was repeated 3 times (Jin et al. 2019). Table S1 provides the primer sequences' details.
Western blotDifferent groups of cells and tissues were collected and individually added to RIPA lysis buffer (P0013B, Beyotime, Shanghai, China) with 1% PMSF on ice for 30 min. The samples were centrifuged at 14,000 g and 4 ℃, and the supernatant was collected. The protein extraction solution was utilized to measure the protein concentration of the samples using the BCA method (P0012S, Bi Yun Tian, Shanghai, China). Add an appropriate 5 × loading buffer and boil at 100 °C for 10 min to denature the proteins. The protein loading amount is 50 μg. First, separate the gel for electrophoresis and then concentrate it. Next, transfer the bands that contain the target protein to a PVDF membrane after electrophoresis. The PVDF membrane was first immersed in a solution containing 5% skim milk powder and then sealed at room temperature for 1 h. Subsequently, the membrane was incubated overnight at 4 °C with primary antibodies, including anti-Flag (rabbit; dilution 1:1000, F7425, Sigma-Aldrich, USA), anti-HA (rabbit; dilution 1:1000, #3724, Cell Signaling, USA), anti-SIRT5 (rabbit; dilution 1:2000, 15,122–1-AP, Proteintech, Wuhan Sanying, China), anti-p53 (rabbit; dilution 1:5000, 10,442–1-AP, Proteintech, Wuhan Sanying, China), anti-pRb (rabbit; dilution 1:1000, #8516, Cell Signaling, USA), anti-Cyclin D1 (mouse; dilution 1:5000, 60,186–1-Ig, Proteintech, Wuhan Sanying, China), anti-p27 (rabbit; dilution 1:1000, 25,614–1-AP, Proteintech, Wuhan Sanying, China), anti-GLUT1 (rabbit; dilution 1:1000, #73,015, Cell Signaling, USA), anti-HKII (rabbit; dilution 1:2000, 22,029–1-AP, Proteintech, Wuhan Sanying, China), and anti-β-actin (rabbit; dilution 1:1000, ab8226, Abcam, UK). β-actin was used as the internal reference. Afterward, the membrane was washed with PBS-T at room temperature and subsequently incubated with secondary antibodies, which were HRP-labeled goat anti-rabbit/goat anti-mouse IgG (dilution 1:10,000, BA1054/BA1050, Boyade, Wuhan, China), at room temperature for 1 h. Finally, the membrane underwent 6 washes with PBS-T, each lasting 5 min. The ECL reaction solution (AR1172, BoDe, Wuhan, China) should be evenly applied onto the membrane and then subjected to exposure using the Amersham Imager 600 (USA). Then use Image J for grayscale analysis (Salem et al. 2019; Shu et al. 2011). The experiment is repeated three times.
Construct a colitis-associated colon cancer (CAC) mouse modelMale p53 gene knockout mice (6 weeks old; p53-/-; on a C57BL6 background strain) were purchased from Saibaio (C001203). Wild-type (WT) male C57BL6 mice, aged 6 weeks, were obtained from Vital River, Beijing, China. The animals were kept in SPF-level animal rooms with a constant humidity of 45%-50% and a temperature range of 25 ~ 27 ℃ for one week. Each day, they were exposed to a 12-h light and 12-h dark cycle to adapt to the experimental environment.
The wild-type (WT) and p53 knockout (p53-/-) mice were assigned to four groups, namely the WT group, p53-/- group, LEVs + WT group, and LEVs + p53-/- group, each consisting of 6 mice. To establish the CAC model, wild-type (WT) and p53-/- mice were intraperitoneally injected with a single dose of azoxymethane (AOM) at 10 mg/kg (Sigma-Aldrich, St Louis, MO, A5486). After five days, a 2.5% solution of dextran sulfate sodium (DSS) was injected into the drinking water for five days. It was followed by 14 days of regular drinking water. After the last administration of 2.5% DSS, 3 weeks elapsed, and then 100 μg (100 μL PBS) of LEVs or PBS (as the control) were injected twice a week via the tail vein. The mice were euthanized 69 days after the AOM injection. Subsequently, the colon mucosa and polyp tissues were dissected for Western blot and immunohistochemistry analysis. Polyp counting and immunohistochemical analysis were conducted by researchers blinded to the treatment methods. The number of polyps is determined by counting the total polyps in a specific mouse. Polyp burden in mice is determined by calculating the sum of the diameters of all polyps (Mao et al. 2022; Neufert et al. 2007).
ImmunohistochemistryThe expression of proliferation-related markers Ki67, Cyclin D1, and p27 was detected in mouse colon polyp tissue using the streptavidin peroxidase (SP) immunohistochemical staining method. Moreover, the expression of SIRT5 was examined in both colorectal cancer tissue and adjacent normal tissue of colorectal cancer patients. Colonic polyp tissue and colon cancer tissue paraffin specimens were obtained from patients. After routine dehydration processing, consecutive sections with a thickness of 5 μm were performed. Subsequently, the specimens were stained using the conventional procedure of immunohistochemistry. To block endogenous peroxidases, the samples were treated with 3% hydrogen peroxide at room temperature for 10 min and then subjected to a 10-min blocking step using normal goat serum. The primary antibodies used were rabbit anti-Ki67 (1:2000, 28,074–1-AP, Proteintech, Wuhan, China), mouse anti-Cyclin D1 (1:500, 60,186–1-Ig, Proteintech, Wuhan, China), rabbit anti-p27 (1:200, 25,614–1-AP, Proteintech, Wuhan, China), and rabbit anti-SIRT5 (1:200, 15,122–1-AP, Proteintech, Wuhan, China). The incubation was carried out overnight at 4 ºC. Following this, the tissue sections were incubated with biotinylated secondary antibodies (Sheep anti-Mouse/Sheep anti-Rabbit, diluted 1:500, BA1001/BA1003, Boster, Wuhan, China) at 37 ºC for 20 min. Subsequently, 50μL of streptavidin-peroxidase solution was added and incubated at room temperature for 10 min. Stain the slides using DAB, followed by restaining with hematoxylin. Finally, dehydrate, clear, and mount the slides for microscopic observation. Using an isotype control instead of PBS is recommended as a negative control.
The criteria used to determine protein-positive cells are as follows: standard positive cells display a brownish-yellow color, and scoring is based on the extent and intensity of staining. The percentage of positively stained area assesses the staining range: score 0 signifies < 5%, score 1 signifies 5–25%, score 2 signifies 25–50%, score 3 signifies 50–75%, and score 4 signifies > 75%. The staining intensity is classified into four levels: 0, 1, 2, and 3. Each number corresponds to a specific staining intensity: 0 represents negative (no staining), 1 represents mild (weak) staining, 2 represents moderate staining, and 3 represents intense (strong) staining. Next, the staining intensity should be multiplied by the percentage of area to calculate the weighted score (Li et al. 2016; Tian and Yuan 2018).
Patient and sample preparationA total of 58 fresh colon cancer tissues and their matched adjacent non-tumor tissues were collected from colon cancer patients who underwent curative colon resection surgery at our hospital between 2017 and 2018, without receiving any chemotherapy or radiotherapy. Among them, there were 33 male and 25 female patients, with a mean age of 55.97 ± 9.24. Nineteen cases had lymph node invasion, while 39 cases did not; 39 cases were classified as TNM stage I-II, and 19 cases as stage III. The 58 colon cancer patient tissues were divided into high and low expression groups based on the median expression levels of SIRT5 and p53 succinylation proteins. Survival analysis was conducted on patients with high and low expression of SIRT5 and p53 succinylation proteins, with a follow-up period of up to 5 years. (Tian and Yuan 2018).
留言 (0)