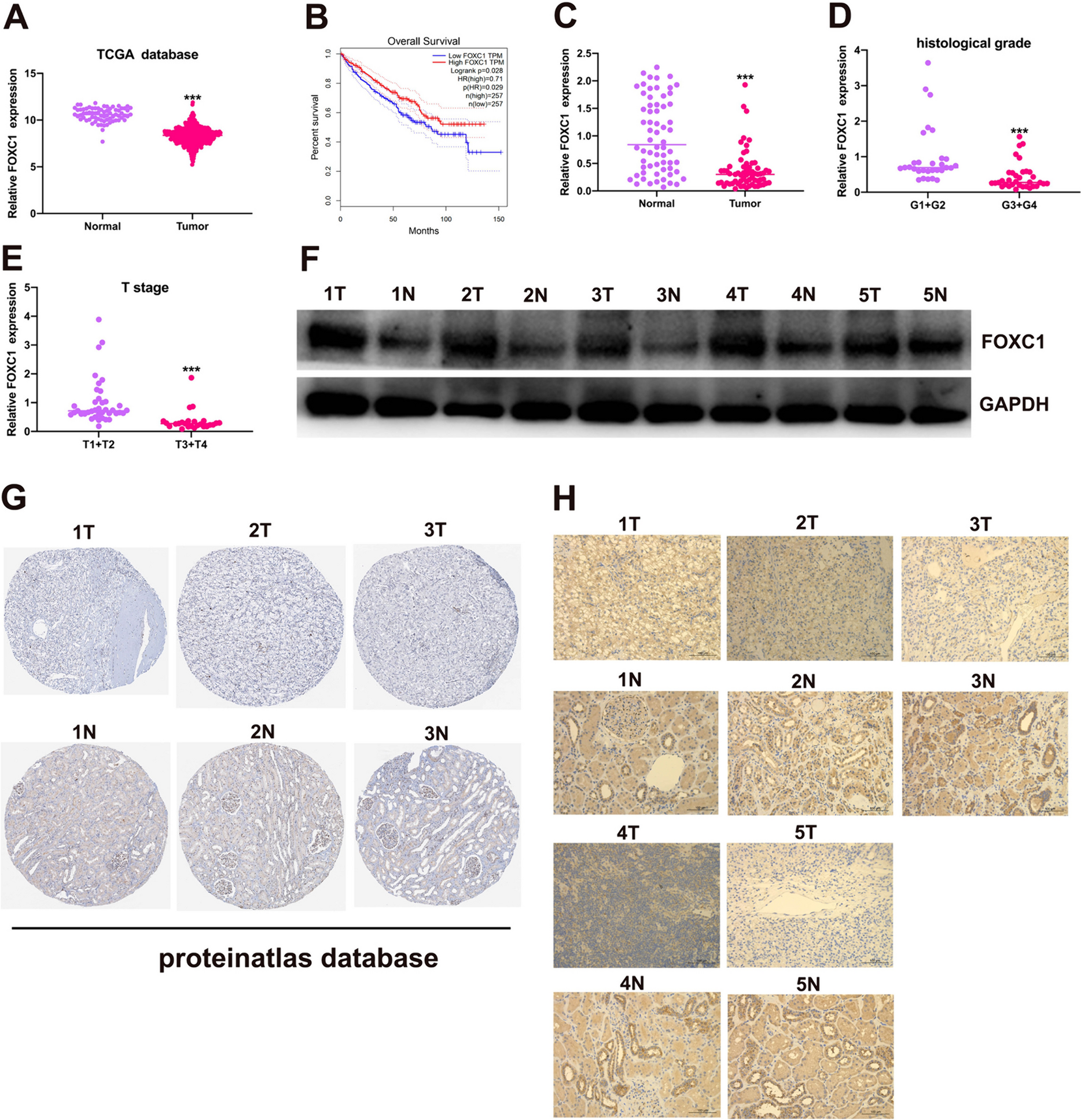
Patients and tissue specimens
This study was approved by the research ethics committees of Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College (NCC2020A190), Sun Yat-Sen University Cancer Center and Shanxi Bethune Hospital affiliated with Shanxi Academy of Medical Sciences. All lung cancer samples were collected with informed consent. Patients admitted to the hospital with a diagnosis of lung cancer by at least two independent pathologists were included in this study. Samples with fresh normal lung tissues (5 cm away from tumor lesions) were obtained from surgical specimens resected during the surgery and immediately frozen in liquid nitrogen for further analysis. A total of 45 frozen samples were obtained to detect the expression of PD-L2 in human lung tumor specimens and the adjacent normal lung tissues, while 25 formalin-fixed and paraffin embedded (FFPE) specimens according to manufacturer’s protocol were collected from lung cancer patients receiving nivolumab immunotherapy to determine the correlation of PD-L2 and FOXP3 expression levels.
Cell lines and cell culture
Human lung cancer cell lines (H1299, H1975, H460, H520, H446, and H82), murine cancer cell lines (LLC, MC-38, and Ag104Ld), and human embryonic kidney line (HEK293T) were purchased from the American Tissue Culture Collection (ATCC; Manassas, VA, USA), while the human normal bronchial epithelial cell line 16HBE was obtained from Lonza Clonetics (Clonetics, Walkersville, MD, USA), and human embryonic lung fibroblast cell line HLF was purchased from Kenqiang Instrument Co., Ltd (Shanghai, China). These cells were cultured in Dulbecco Modified Eagle Medium (DMEM) or Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified incubator at 37°C with 5% CO2.
Antibodies and reagents
The antibodies used in this study were as follows: rabbit anti-human PD-L2 (#82723, Cell Signaling Technology, Beverly, MA, USA; 1:100 for immunohistochemistry (IHC)), rabbit anti-human PD-L1 (#13684, Cell Signaling Technology, Beverly, MA, USA; 1:100 for IHC), rabbit anti-human FOXP3 (#98377, Cell Signaling Technology, Beverly, MA, USA; 1:100 for IHC), anti-p-p65 (#3033, Cell Signaling Technology, Beverly, MA, USA; 1:1000 for western blot), anti-PD-L2 (#ab87662, Abcam, Cambridge, MA, USA; 1:1000 for western blot), mouse anti-PD-L2 (#bsm-30107M, Bioss antibodies, Beijing, China), rabbit anti-RGMB (#bs-11474R, Bioss antibodies, Beijing, China; 1:1000 for western blot), anti-RGMB (#A12859, Abclonal, China; 1:1000 for western blot), anti-PD-L2 (#ab288298, Abcam, Cambridge, MA, USA; 1:50 for Co-immunoprecipitation); anti-β-Actin (#A1978, Sigma, St. Louis, MO, USA; 1:5000 for western blot), anti-PD-L2 (#sc-57398, Santa cruz, USA; 1:50 for IHC), anti-PD-L2 (#NBP1-76770, Novus iologicals, LLC, USA; 1:100 for immunofluorescence assay), anti-p65 (#A19653, Abclonal, China; 1:1000 for western blot), anti-AhR (#83200, Cell Signaling Technology, Beverly, MA, USA; 1:1000 for western blot, 1:50 for Chromatin immunoprecipitation (CHIP)), Zombie Aqua™ dye (#423101,Biolegend; 1:1000 for flow cytometry), BUV395 anti-mouse CD45 (#745698, BD OptiBuild, 1:100 for flow cytometry), APC anti-mouse CD4 (#100411,Biolegend; 1:20 for flow cytometry), PE/Cy7 anti-mouse CD8a (#100721,Biolegend; 1:20 for flow cytometry), APC/Cy7 anti-mouse CD11b (#101225, Biolegend; 1:20 for flow cytometry), PE/Cy5 anti-mouse F4/80 (#123111, Biolegend; 1:20 for flow cytometry), PE anti-mouse Gr1 (#108407, Biolegend; 1:20 for flow cytometry), APC anti-mouse CD206 (#141707, Biolegend; 1:20 for flow cytometry), PE/Cy7 anti-mouse MHCII (#107629, Biolegend; 1:20 for flow cytometry), PE/Cy7 anti-mouse CD86 (#105013, Biolegend; 1:20 for flow cytometry), PE anti-mouse FOXP3 (#12-4776-41, eBioscience; 1:20 for flow cytometry), PE anti-human PD-L1 (#329705, Biolegend; 1:20 for flow cytometry), and PE anti-human PD-L2 (#329605, Biolegend; 1:20 for flow cytometry). Benzo(a)pyrene (#B1760), Alpha-Naphthoflavone (ANF; #N5757), Puromycin, and RNase A were purchased from Sigma-Aldrich. PD98059 was purchased from Selleck Chemicals.
Flow cytometry analysis
The 16HBE, HLF, H446, H82, H1975, H460, H520, LLC, MC-38, and Ag104Ld cells with or without IFN-γ treatment were collected and incubated with PE-conjugated anti-PD-L2 (#329605, Biolegend) or PE-conjugated anti-PD-L1 (#329705, Biolegend) on ice. The cells incubated with isotype control was set as negative control. After 30 min incubation, the cells were washed twice with PBS, and subjected for flow cytometry analysis by using BD Fortessa 421 (LSRFortessa X-20, BD, USA).
RNA extraction and quantitative real-time PCR (qRT-PCR)
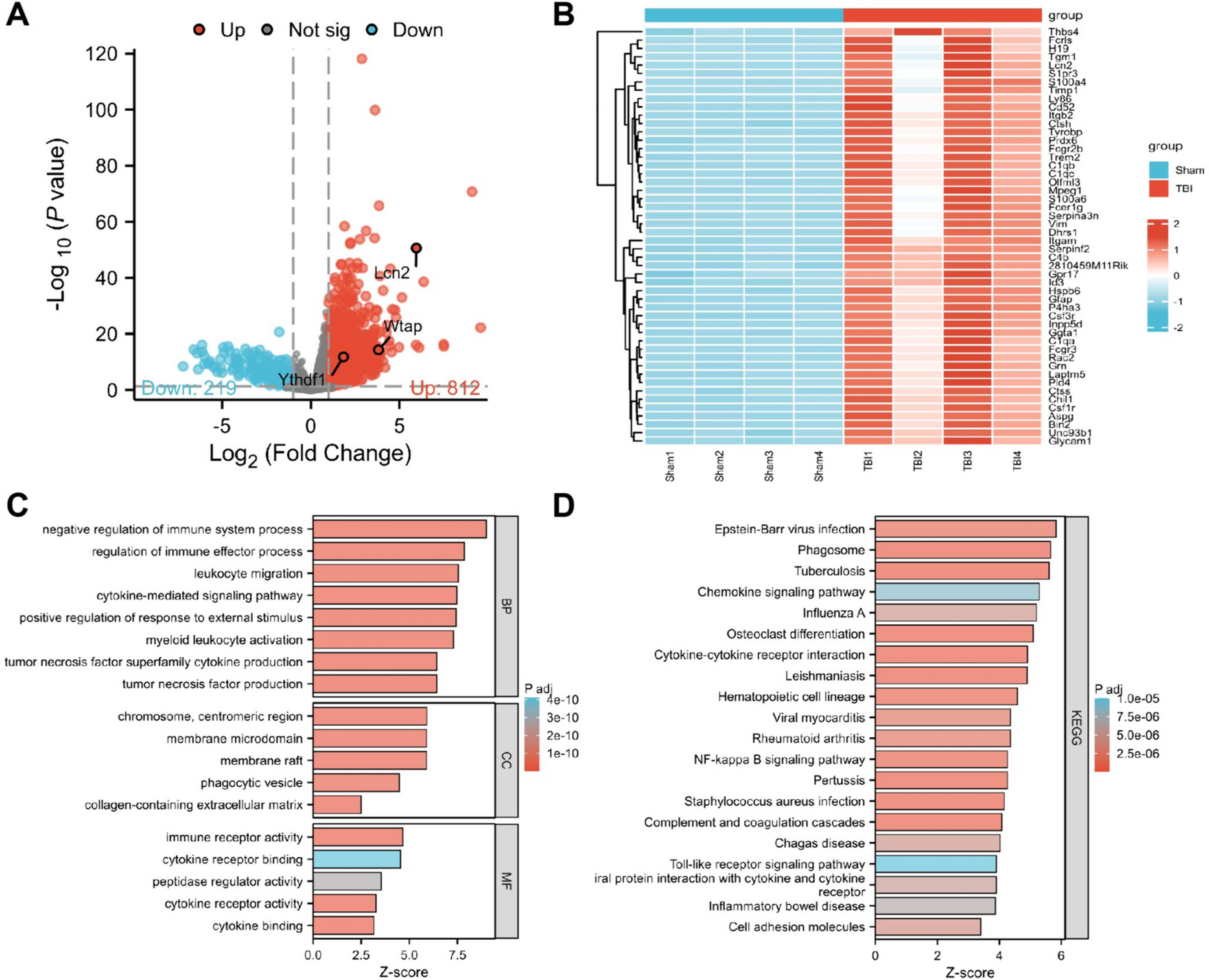
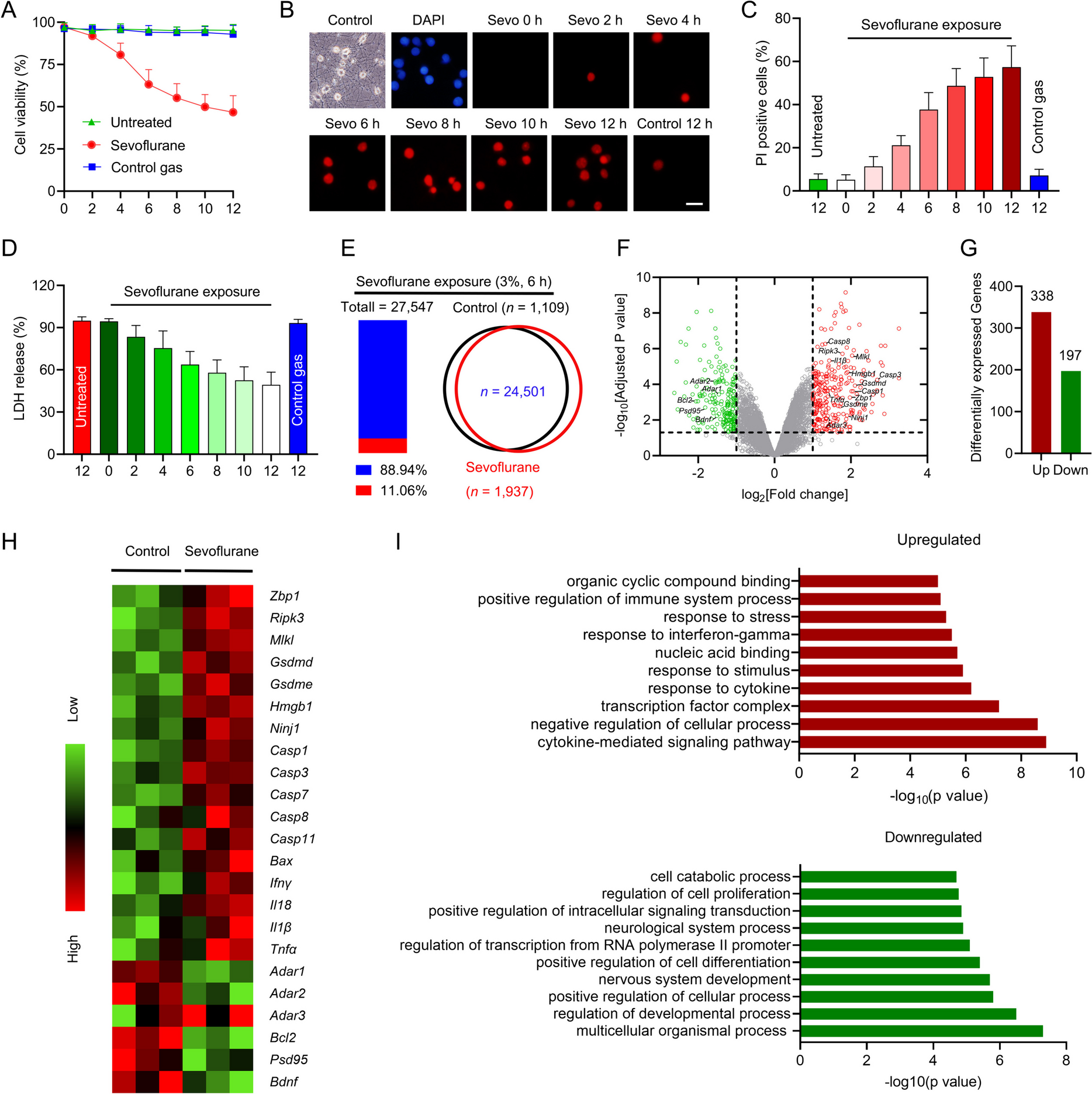
Total RNA was extracted from cells using the Trizol Reagent (Invitrogen, Frederick, MD, USA), and the concentration of isolated RNA was measured by using a NanoDrop spectrophotometer 2000 (Thermo Fisher Scientific). Then 2 μg of RNA was reverse-transcribed into cDNA using a 1st-STRAND cDNA Synthesis Kit (Fermentas, Pittsburgh, PA, USA) in accordance with the manufacturer’s protocol. Follow the transcription, qRT-PCR was performed using SYBR PremixExTaq (Takara Biotechnology, Dalian, China). The sequences of the primers used were listed in Supplementary Table 1. GAPDH was used as an internal reference gene, and the relative expression of genes was calculated using the 2−ΔΔCt method. The sequences of siRNAs, shRNAs, and sgRNAs were listed in Supplementary Table 1.
Immunohistochemistry analysis
Briefly, formalin-fixed, paraffin-embedded human or mouse lung cancer tissue specimens (5 μm) were deparaffinized using xylene and graded alcohol, and then subjected to a heat-induced epitope retrieval step in citrate buffer solution. Then sections were blocked with 5% BSA for 30 min, and incubated with indicated primary antibodies at 4°C overnight. After that, sections were incubated with secondary antibodies for 90 min at 37°C, and then stained with 3, 30-diaminobenzidine (DAB, Zhongshan Golden Bridge Biotechnology, Beijing, China). Three fields of view per sample were imaged and analyzed. Immunoreactivity scores (IRS) were calculated by IRS (0–12) = RP (0–4) × SI (0–3), where RP is the percentage of positive cells and SI is staining intensity. For the multiplex immunohistochemistry assay, the PANO 4-plex IHC kit (PN130721AD, Panovue, Beijing, China) was used according to the manufacturer’s instructions. Specimens were incubated with anti-FOXP3 (#98377, Cell Signaling Technology) and anti-PD-L2 (#82723, Cell Signaling Technology). Slides were scanned and analyzed using the PerkinElmer Mantra Quantitative Pathology Imaging System (Waltham, Massachusetts, USA).
Immunofluorescence microscopy
Cells were plated on the coverslips (24 mm x 24 mm) and then fixed with 4% paraformaldehyde for 15 min. After permeabilized with 0.3% Triton X-100 in PBS for 20 min and blocked with 5% BSA, the cells were incubated with indicated primary antibodies overnight at 4°C. Then the cells were subjected to PE-labeled secondary antibody or Alexa Flour® 488/647-labeled secondary antibody (Life technologies) in PBS for 2 h. The resulting cells were imaged using a laser scanning confocal microscope (Zeiss, Oberkochen, Germany).
Lentivirus-mediated transfection
To generate PD-L2 stable knockdown or overexpression cells, the shRNA sequences targeting human PD-L2 were inserted into a PLKO.1 vector to construct the PLKO.1/shPD-L2 expression vector, while the cDNA sequences of PD-L2 were subcloned into pCDH-GFP vector to construct the pCDH-GFP/PD-L2 expression vector. Then these vectors were co-transfected with psPAX2 and pMD2G into HEK293T cells. After 6 h transfection, the culture medium was replaced with fresh medium, and the lentiviral vector-containing supernatants were harvested 48 h and 72 h post transfection and centrifuged to obtain lentiviral particles. Targeted cells were infected with lentiviral particles in the presence of 8 μg/mL polybrene, and expression of GFP was used as a quantitative measure of infection using a MoFlo XDP cell sorter (Beckman Coulter).
Enzyme-linked immunosorbent assay (ELISA) and Co-immunoprecipitation (Co-IP)
Cells were plated in the 6-well plates at a density of 1x105 cells per well and incubated at 37°C for 48 h. Then the supernatants from each well were collected, and the CCL20 concentration in culture supernatants of lung cancer cell lines was measured by ELISA kit (#441404, Biolegend, USA) as previously described (Lian et al. 2021). The samples were diluted 2-fold before CCL20 ELISA assay analysis. For Co-IP, the PD-L2-overexpressed cells were cultured in a 100 mm-culture dish and lysed in immunoprecipitation lysis buffer containing phosphatase inhibitors and protease inhibitors. The cell lysate supernatant was obtained via centrifugation at 4°C (12,000 g, 15 min), and then incubated with protein A/G-linked magnetic beads (Santa Cruz, CA, USA) and anti-PD-L2 antibody at 4°C overnight. The immunoprecipitants were washed 5 times with immunoprecipitation lysis buffer and boiled with 2xSDS loading buffer. After that, the Co-IP samples were subjected to western blot analysis.
Proximity ligation assay (PLA)
H1975 and H460 cells were seeded at a 6-well plate and transfected with exogenous PD-L2 plasmid. After 24 h of transfection, cells were fixed in 4% paraformaldehyde for 30 min and washed with PBS. The proximity of PD-L2 to RGMB was assessed using the Duolink® In Situ PLA® Probe Anti-Rabbit PLUS, Duolink® In Situ PLA® Probe Anti-Mouse MINUS, and Duolink® PLA In Situ Detection Reagents Red (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Specifically, the rabbit anti-RGMB antibody (1:200) and mouse anti-PD-L2 antibody (1:200) were used for PLA assay. Fluorescent images were taken using a laser scanning confocal microscope.
Isolation of lymphocytes
Human peripheral blood mononuclear cells (PBMCs) were isolated from the heparinized peripheral blood of healthy volunteers by Ficoll density gradient-based separation. Then human CD4 Microbeads (Miltenyi Biotec, catalog 130-045-101) were used to isolate CD4+ T cells from the PBMCs according to the manufacturer’s instructions. CD4+CD25+ Tregs were further sorted using flow sorting.
Transwell migration assays
Transwell migration assays were carried out in 24-well plates with inserts (5 μm, Corning Incorporated, Corning, NY, USA) according to the manufacturer’s instructions. Briefly, Tregs (1 × 105 cells/well) or PBMCs (2 × 105 cells/well) were plated to the upper chamber, and 600 μL RPMI-1640 medium containing recombinant human CCL20 (10 ng/ml; Peprotech, USA), anti-CCL20 antibody (1 ng/ml; AF360-SP, R&D, USA), supernatants taken at 24 h from pure cultures of PD-L2-overexpressed cells or shPD-L2 transfected cells were added to the lower chambers. After 24 h incubation, cells migrated to the lower chamber were manually counted.
Western blot analysis
The cells were lysed in RIPA lysis buffer (50 mM Tris-HCl (pH 7.5), 150mM NaCl, 1mM EDTA, 1 mM MgCl2, 0.5% Triton X-100) supplemented with phosphatase inhibitors and protease inhibitors. Protein concentration was determined by using the BCA method (Biyuntian, China). Equal amount of protein (50 μg) from each sample was loaded and separated on a 10% SDS-PAGE gel, and then transferred to nitrocellulose blotting membranes (GE Healthcare Life science). After blocking with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST), membranes were incubated with the indicated primary antibodies overnight at 4°C. Then membranes were washed with TBST and incubated with the corresponding secondary antibodies at room temperature. GAPDH and β-actin were used as internal controls. The immunocomplex on the membrane was visualized using Luminescent Image Analyzer LSA 4000 (GE, Fairfield, CO, USA).
Luciferase-based reporter assay
The H460 cells were seeded in 12 well plates and incubated overnight for adhesion. After reaching 60% confluence, cells were co-transfected with Firefly luciferase plasmid under the control of wild-type or mutant PD-L2 promoter and Renilla luciferase plasmid for 6 h. Then the medium was refreshed and cells were treated with BaP for additional 48 h. After that, cells were lysed in lysis buffer for 15 min, and the supernatant was collected for luciferase activity measurement. Firefly and Renilla luciferase activities were measured by a dual-luciferase reporter assay system (Promega, Madison, WI) according to the manufacturer’s instructions. The PD-L2 promoter activity was expressed as ratio of Firefly luciferase to Renilla luciferase.
Chromatin immunoprecipitation (ChIP) assay
The H460 cells were treated or untreated with 5 μM BaP for 48 h, collected and fixed with 1% formaldehyde. Then the cell pellets were lysed in RIPA lysis buffer and sheared using sonication. The specific protein/DNA complexes were immunoprecipitated using an antibody against AhR (#83200, Cell Signaling Technology, 1:50). Following immunoprecipitation, cross-linking was reversed, the residual RNA was digested by RNase A (10 μg/mL), while the proteins were removed by proteinase K (40 μg/mL). The resulting DNA was purified and analyzed by quantitative PCR (qPCR). The primers used to detect XRE1 were listed in Supplementary Table 1.
CRISPR/Cas9 assay
The CRISPR/Cas9 assay was performed as previously described by Feng Zhang, et al (Ran et al. 2013). Briefly, the lentiCRISPR plasmid pX330 obtained from Addgene was digested by BsmB1. The pX330-sgAhR plasmids expressing human Cas9 and sgAhR were prepared by ligating sgAhR oligonucleotides into the BsmB1 site of pX330. The sgRNA target sequence for AhR was listed in Supplementary Table 1. Then the H520 cells were transfected with constructed plasmids for 48 h. The single positive cells were sorted by fluorescence activated cell sorting (FACS) into 96-well plates. After three weeks after single-cell sorting, cells were collected to determine the knock-out efficacy by sequencing and western blot analyses.
Animal study
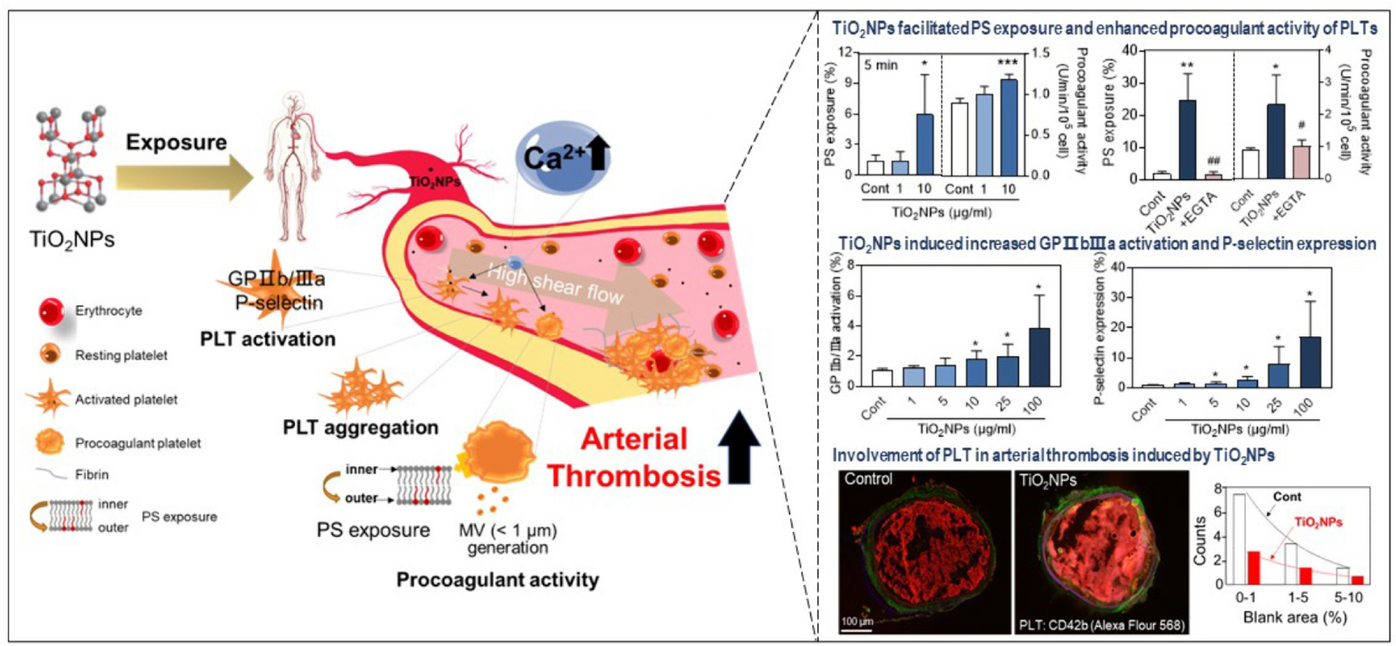
The animal studies were approved by the Institutional Review Board of Institute of Zoology, Chinese Academy of Sciences, and were conducted according to protocols approved by the Animal Ethics Committee of the Institute of Zoology, Chinese Academy of Sciences. Female C57BL/6 mice (5-6 weeks old) were purchased from the Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). Female A/J mice (5-6 weeks old) and homozygous AhR-deficient mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). The A/J mice were exposed to cigarette smoke or treated with BaP to develop lung tumors as previously described (Wang et al. 2019). Briefly. The A/J mice (n = 7 per group) were exposed to cigarette smoke generated by DSI’s Buxco Smoke Generator (Buxco, NC, USA) at a frequency of 12 cigarettes per day, 5 days per week for 60 days. The duration of cigarette smoke exposure per cigarette was 3 minutes, followed by a 15-minute fresh air exposure. In other settings, the A/J mice (n = 7 per group) were orally administrated with 100 mg/kg of BaP twice a week for 5 weeks. The A/J mice were sacrificed on the 60th day after administration for further experiments. The 60-day duration was to make the likelihood of lung tumorigenesis in mice higher (Wang et al. 2019). The C57BL/6 mice were injected with LLC cells (5 ×105) via tail vein to establish lung cancer mouse model or subcutaneously implanted with 5 ×105 of MC-38 cells. Tumor growth was monitored by micro-CT (Quantum FX, PerkinElmer, USA) for LLC mouse model or measured using a digital Vernier caliper for MC-38 mouse model. The tumor volume for MC-38 mouse model was calculated by the formula: volume (mm3) = (width)2 × length /2. At the end of the experiment, all animals were sacrificed with CO2 overdose before cervical dislocations and tumor or lung tissues were collected, weighted, and stored for further experiments.
Ex vivo flow cytometry analysis
The mouse lung tissues were dissected into 2 mm pieces, and digested by collagenase IV (0.3%; Sigma) at 37 °C for 1 h. A single-cell suspension was obtained through a 70 μm cell strainer (BD Falcon, BD Biosciences, USA). The cells were then centrifugated and resuspended in ACK lysis buffer (Beyotime Biotechnology, China) for erythrocyte removal. After that, the resulting cells were labeled with indicated antibodies (BioLegend) and sorted by BD Fortessa 421. The data were analyzed on FlowJo (BD). The Zombie Aqua™ dye was used to determine live cells. The Foxp3+ cells gating on live CD45+CD3+CD4+ T cells represented Treg cell, the MHCII+ cells gating on live CD45+Gr1+ cells represented MDSC cell, the CD86+ cells gating on CD45+CD11b+F4/80+ cells represented M1 cell, and the CD206+ cells gating on live CD45+CD11b+F4/80+ cells represented TAM cell. Matched isotype controls were used in all experiments.
Statistical analyses
All experiments were carried out at least in triplicate. The statistical analyses were performed using Graph Pad Prism 8 software (Graph Pad Software, La Jolla, CA, USA). Data obtained from different groups were compared using Student’s t test, chi-squared test, or one-way analysis of variance (ANOVA). The log-rank test was applied for survival analysis. Spearman correlation analysis was performed to determine the relationship between variables. Fisher exact test was used for the correlation comparison of characteristics of the NSCLC patient. P < 0.05 was considered statistically significant.



留言 (0)