Subjects
From January 2021 to December 2022, 200 AR patients diagnosed in the Otolaryngology Clinic of Ordos Central Hospital were selected as the AR group, and 50 healthy people who underwent physical examination in the hospital during the same period were randomly selected as the healthy control (HC) group. The levels of GATA-3mRNA, RORγtmRNA and FoxP3mRNA in peripheral blood mononuclear cells were detected by real-time fluorescence quantitative PCR (qRT-PCR). The proportions of Th2, Th17 and Treg cells were detected by flow cytometry. The concentrations of IL-4, IL-5, IL-17 and IL-10 in serum were detected by enzyme-linked immunosorbent assay.
The inclusion criteria for the healthy control group were as follows: allergen skin prick test-negative and serum allergen-specific IgE-negative, no specific immunotherapy or glucocorticoid therapy before the experiment, and signed informed consent.
The inclusion criteria for the AR group were as follows: All AR patients met the diagnostic criteria in the literature [1], with 2 or more of the symptoms including sneezing, watery discharge, nasal itching and nasal congestion, which lasted or were accumulative for more than 1 h every day, or accompanied by ocular symptoms such as itching and tearing. Additionally, the patients exhibited signs of nasal mucosal edema and watery nasal secretions upon physical examination. The diagnosis of Artemisia allergy was confirmed by experienced clinicians based on a positive allergen inhalation test (AIT) for Artemisia allergens, indicating sensitivity to these specific allergens, as well as positive serum-specific IgE tests against Artemisia allergens. The presence of subjective symptoms as described above, along with positive AIT and IgE tests, established the diagnosis of AR.
The exclusion criteria: Medical history inquiry showed no upper respiratory tract infection within 2 weeks, and excluded bronchiectasis, autoimmune diseases and allergic asthma; patients received specific immunotherapy and glucocorticoid therapy in the past month; patients were complicated with other nasal diseases, or severe lesions of the heart, liver, kidney and other organs; patients were unwilling to participate in this study.
The inclusion criteria for healthy individuals included: allergen skin prick test-negative and serum allergen-specific IgE-negative, no specific immunotherapy or glucocorticoid therapy before the experiment, and signed informed consent.
This study was approved by the Ethics Committee of Ordos Central Hospital (ethical approval No.: 2021-030), and all subjects signed the informed consent.
MethodsCollection of blood samples
Three tubes of fasting peripheral venous blood were aseptically collected from confirmed AR patients and healthy controls in the morning of the second day, including 5 ml blood in an anticoagulant tube containing heparin sodium, 2 ml blood in an EDTA-K2 anticoagulant tube, and 3 ml blood in a separating gel coagulation-promoting tube. After anticoagulant blood collection, repeated reversal was needed 8–10 times for full mixing. All blood samples were processed within 2 h after collection.
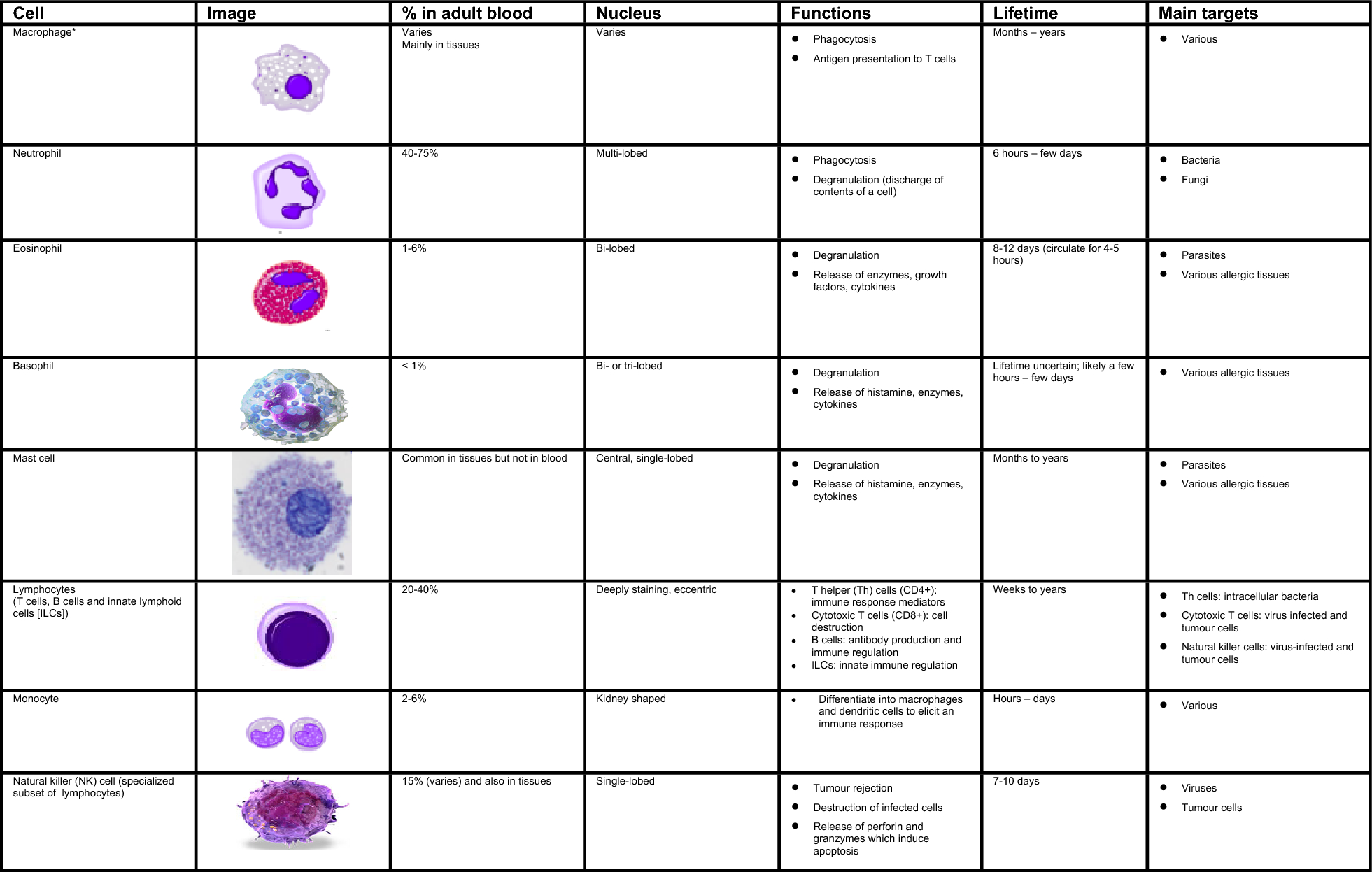
Detection of Th2, Th17 and Treg cell proportions in peripheral blood
Two Falcon tubes were added with 200 µl heparin sodium anticoagulant whole blood and 4 µl stimulant, respectively, followed by even mixing and incubation in a water bath at 37℃ for 6 h. CD4 FITC fluorescent antibody (20 µl) was added to each tube, which was mixed well and incubated at room temperature in the dark for 15 min. Perm/wash buffer I (100 µl) was added and mixed well for incubation at room temperature in the dark for 5 min. 1X BD FACS lysing solution (2 ml) was added and well mixed, followed by incubation at room temperature in the dark for 10 min, and centrifugation at 800 g for 5 min. After discarding the supernatant, 50 µl perm/wash buffer II was added. One tube was added with 20 µl IL-4 PE fluorescent antibody, and the other with 20 µl IL-17 A PE fluorescent antibody, subjected to mixing well and incubation at room temperature in the dark for 30 min. After washing the cells with PBS, 500 µl PBS was added to each tube and mixed well. The proportions of Th2 and Th17 cells were analyzed using BD FACSCalibur flow cytometry. During the experiment, controls of the same type were set, with CD4+IL-4+ as control for Th2 cells and CD4+IL-17+ for Th17 cells.
A Falcon tube was added with 100 µl heparin sodium anticoagulant whole blood, and then added with 20 µl CD25 APC fluorescent antibody and 20 µl CD4 FITC fluorescent antibody, followed by well mixing and incubation at room temperature in the dark for 15 min. 1X BD FACS lysing solution (2 ml) was added and well mixed for red blood cell lysis at room temperature in the dark for 10 min. After washing cells twice with PBS, the supernatant was discarded and the freshly configured FoxP3 Fix/Perm buffer was added. The cells were incubated at room temperature in the dark for 40 min for fixation, and then perm/wash buffer was added to break the cell membrane. After washing the cells with PBS, 20 µl FoxP3 PE fluorescent antibody was added and mixed well, followed by staining at room temperature in the dark for 40 min. After cell washing with PBS, 500 µl PBS was added and mixed well, and the proportion of Treg cells was analyzed by BD FACSCalibur flow cytometer. During the experiment, controls of the same type were set, with CD4+CD25+FoxP3+ as control for Treg cells.
Extraction of total RNA from peripheral blood mononuclear cells
Heparin sodium anticoagulant blood (4 ml) was taken and added with 4 ml 0.01 M PBS, which was mixed well and then slowly added into a centrifuge tube containing 4 ml lymphocyte separation medium. After centrifugation at 2,000 rpm/min at room temperature for 15 min, the white cloudy layer dominated by mononuclear cells was sucked into a 10-ml centrifuge tube, which was added with 5 ml 0.01 M PBS and mixed well for centrifugation at 1,500 rpm/min for 10 min and supernatant discarding. After repeated washing twice, mononuclear cells were collected into a 1.5-ml EP tube. Total RNA was extracted from mononuclear cells according to the instructions of the total RNA extraction kit of Tiangen Biotech (Beijing) Co., Ltd. After extraction, the concentration and purity of RNA were determined using an ultraviolet (UV) spectrophotometer, and then the RNA samples were cryopreserved in a refrigerator at -80℃ for next use.
Detection of GATA-3, RORγt and FoxP3 mRNA levels
With total RNA of mononuclear cells as template and GAPDH as internal reference gene (forward primer: 5’CAAGGCTGTGGGCAAGGTCATC-3’, reverse primer: 5’-GTGTCGCTGTTGAAGTCAGAGGAG-3’), the reaction system was added according to the instructions of the FastKing one-step RT-PCR kit produced by Tiangen Biotech (Beijing) Co., Ltd. for reverse transcription (42℃ 30 min, 95℃ 3 min) and RT-PCR (94℃ 30 s, 65℃ 30s, 72℃ 30 s, 40 cycles, 72℃ 5 min). ABI 7500 real-time fluorescent quantitative PCR instrument was used for detecting the levels of GATA-3 mRNA (forward primer: 5’-CAGTTGGCCTAAGGTGGTT-3’, reverse primer: 5’-GCACGCTGGTAGCTCATACA-3’), RORγt mRNA (forward primer: 5’-GCTGTGATCTTGCCCAGAACC-3’, reverse primer: 5’-CTGCCCATCATTGCTGTTAATCC-3’), and FoxP3 mRNA (forward primer: 5’-CTGCCCCTAGTCATGGTGG-3’, reverse primer: 5’-CTGGAGGAGTGCCTGTAAGTG-3’). Replicates were made for each sample. After reaction, the melting curve was drawn to distinguish specific and non-specific amplification, and the relative expressions of target genes were calculated by the 2–ΔΔCT method.
Count of eosinophils and basophils in peripheral blood
Whole blood cell count was carried out using EDTA-K2 anticoagulant blood. The internal quality control of samples was conducted before detection. All detection items were tested by BC-7500CS full-automatic blood cell counter and its supporting reagents within 1 h after blood collection.
1.2.6 Detection of serum cytokines IL-4, IL-5, IL-17, IL-10 and IgE.
After collection by coagulation-promoting tube, blood was placed at room temperature for 30 min. After coagulation, blood was centrifuged at 3,500 r/min for 10 min (with a centrifugal radius of 10 cm), and the serum was divided into two parts. One part was used to detect the concentration of IgE by E601 fully automatic electrochemiluminescence immunoassay analyzer and its supporting reagents, with internal quality control before sample detection. The other part was used for detection of cytokines IL-4, IL-5, IL-17 and IL-10 using an ELISA kit according to the instructions. OD value was measured at 450 nm by a microplate reader, and cytokine concentrations were calculated according to the standard curve.
Main instruments and reagents
The main instruments and reagents used are as follows: BC-7500CS full-automatic blood cell counter (Mindray, China), E601 fully automated electrochemiluminescence immunoassay analyzer (Roche, USA), BD FACSCalibur flow cytometer (BD, USA), ABI 7500 real-time fluorescent quantitative PCR instrument (Thermo Fisher, USA), DW-86L388 -80℃ ultra-low temperature refrigerator (Haier, China), lysing solution, perm/wash buffer, stimulant and IL-4/IL-17 A/CD25/CD4/FoxP3 fluorescent antibody (BD, USA), RNA Easy Fast total RNA extraction kit and FastKing one-step RT-PCR kit (Tiangen Biotech Co., Ltd., Beijing), and human IL-4, IL-5, IL-10, IL-17 ELISA kit (NeoBioscience Technology Co., Ltd., Shenzhen).
Statistical methods
The data were analyzed using SPSS 27.0. The counting data were expressed as rate, and their inter-group comparisons were conducted by the χ2 test. The measurement data conforming to the normal distribution were expressed as \(\overline x + s\) and compared between groups using the t test or corrected t-test, and those of non-normal distribution were expressed as median and quartile [M (P25 ∼ P75)] and compared between groups with the nonparametric test. The correlation between the two groups was analyzed by linear regression. P < 0.05 was considered as statistically significant.


留言 (0)