Ethics statement
Animals were used in compliance with the Animal (Scientific Procedures) Act 1986, and with approval from St. George’s University of London (SGUL), London School of Hygiene and Tropical Medicine (LSHTM) and University of Leicester (UoL) Ethics approval under Home Office animal project licenses 70/7490 and P6DCE1A76 was obtained. The study was carried out in compliance with the ARRIVE guidelines.
Mice and immunisations
All mice used in the experiments were kept under specific pathogen free (SPF) conditions at the Biological Resource Facility at SGUL, Biological Services Facility of LSHTM, and the preclinical resource facility of UoL. All animals were fed with a standard rodent diet and provided with water ad libitum. For LSHTM animal experiments, 6- to 8-week-old BALB/c mice were purchased from Charles River, randomly allocated in groups of 5–6 per cage and acclimatised for 7 days. For SGUL and UoL experiments, BALB/c and C67BL/6 mice, respectively, were sourced from the in-house breeding facility, age-matched and randomly allocated in groups of five to six animals per each cage. Mice were immunised with 100 µL sterile Dulbeco’s phosphate buffered saline (DPBS) or BCG Pasteur (5 × 105 CFUs/mouse) subcutaneously at the base of the tail. After 3 or 7 weeks, euthanasia by cervical dislocation was done and spleens were collected from each mouse.
Culture and use of laboratory strains of mycobacteria
Mtb H37Rv, Mtb H37Rv-luciferase (Mtblux), BCG Pasteur (BCGWT), BCG-luciferase (BCGlux) and BCG expressing green fluorescent protein (BCGGFP) were cultures maintained at the Institute for Infection and Immunity, St. George’s, University of London. H37Rv and BCGWWT were propagated in Difco™ Middlebrook 7H9 Broth (BD, 271391) supplemented with 10% v/v Middlebrook 7H10 oleic acid, albumin, dextrose, and catalase (OADC) (USBiological Life Sciences, M3895-01) and 0.5% v/v glycerol (VWR Chemicals, 56-81-5). Culture media for Mtblux, BCGGFP and BCGlux were supplemented with 50 µg/mL hygromycin B (ChemCruz®, SC-29067) as a selection marker and 0.05% tyloxapol (Sigma, T0307-10G) as detergent to prevent clumping. All mycobacterial cultures were propagated for up to 3 weeks. Frozen stocks were made with sterile deionised distilled water with 10% glycerol kept at −80 °C or −196 °C. The percentage of GFP-expressing BCG in samples was >90% as determined by flow cytometry (data not shown).
To quantify colony-forming units (CFU) in samples, serial dilutions of bacteria were plated on solid media comprising of BBL™ Seven H11 agar base (BD, 212203) supplemented with OADC and Mast® Selectatab (Mast group, MS24). Plates were kept in a sealed bag and left in an incubator set to 37 °C for around 3 weeks. Contamination was monitored by assessing for growth of contaminants for 48–72 h after initial seeding of cultures. All experiments with BCG were performed in Class II biosafety cabinets and experiments with Mtb in Class I biosafety cabinets housed in the CL3 suite of St. George’s University of London.
Luciferase assay
Luciferase assay was done to quantify viable bacteria as previously described38. Briefly, cell lysates were added to tubes (Greiner Bio-One, 115101) with 1 mL of 0.1% n-decyl aldehyde (Decanal) (Sigma-Aldrich, W236217), prepared by doing a 1 in 10 (v/v) dilution of 1% Decanal in PBS. Bioluminescence was measured using a Junior LB 9509 Portable Luminometer (Berthold Technologies GmbH & Co.) set to measure luminescence for 30 s. Data are expressed as relative light units (RLU).
Murine cell lines and cell culture
J77439 (BALB/c peritoneal monocyte cell line) and MH-S40 (BALB/c alveolar macrophage cell line) were obtained from ATCC while the BL/6-M41 (C57Bl/6 bone marrow-derived macrophage cell line) cell line was obtained from Kerafast® (ENH166-FP). RPMI (Sigma, R0833) and DMEM (Sigma, D6546) were supplemented with 10% Foetal Bovine Serum (FBS,) (Sigma-Aldrich, F9665), 5mM L-Glutamine (Sigma, G7513), 100 U/mL Penicillin & 100 µg/mL Streptomycin (Sigma, P4333), 50 µM β-mercaptoethanol (Sigma, M3148), and 10 mM (4-(2-hydroxyethyl)-1-piperazineethanosulfonic acid (HEPES) buffer (Gibco, 15630-056), and used in all assays, except for omission of antibiotics in infection assays. Supplemented DMEM and RPMI-1640 are herein referred to as D10 and R10, respectively. MH-S cells were maintained in R10 (Gibco) while J774 and BL/6-M cells were initially cultured in D10 but were conditioned to be cultured in R10 media since splenocytes used for coculture experiments required the use of R10. There was no significant difference in the viability of J774 and BL/6-M cells after culture in R10 medium. For infection assays, cells were detached by first removing media, then gently washing the cells twice with sterile DPBS (Sigma, D8547). Afterwards, cells were incubated for 5 mins in cell dissociation buffer (EMD Millipore Corp USA, W4502) and harvested by washing the plate with complete medium. After centrifugation at 300 rcf for 5 mins, cells were resuspended in complete medium and counted using an automated cell counter (TC20™, Bio-Rad Laboratories, Inc.) using Trypan Blue exclusion method. All cell cultures were maintained in a humidified CO2 incubator (5% CO2 and 37° C).
Infection of cell lines BCGlux, BCGGFP and Mtblux
MH-S, J774 and BL/6-M cells were seeded in 96- (Corning, 353072) or 48- (Corning, 3548) well plates at a density of 500,000 cells per well. After leaving the cells to settle for 2 h, cells were infected with mycobacteria at different multiplicities of infection (MOI): 1:1, 2:1, and 4:1. Infected cell lines were harvested from multi-well plates by aspirating culture medium, then adding cell dissociation buffer. After 5 mins in the CO2 incubator, fresh medium was added to each well and cells were detached from the bottom of the wells by vigorous pipetting. Cells were transferred to 96-well U bottom plates and washed thrice with sterile PBS prior to FACS staining and analysis. For luciferase assays, cells were directly lysed with 200 µL sterile water (Sigma, 3864580) after aspiration of culture medium.
Mouse splenocyte isolation and culture
Aseptically collected spleens were placed in tubes with MACS® tissue storage solution (Miltenyi Biotec, 130-100-008) and maintained on ice to keep the integrity of the organs prior to processing. Single-cell suspensions were obtained by mechanical maceration of the spleen with a syringe barrel through a 70 µm cell strainer (Falcon, 352350) in a 50 mL Falcon tube. The cell strainer was rinsed with 10 mL complete RPMI-1640, then tubes were centrifuged at 300×g for 5 mins in a cold centrifuge set to 4 °C. After discarding the supernatant, red blood cells (RBCs) were lysed by the addition of 2 mL ACK lysis buffer (Gibco, A10492-01), and incubation at room temperature for 2 mins. Lysis was arrested through the addition of 20 mL R10 medium and tubes were again centrifuged at 300×g for 5 mins. Pelleted cells were resuspended in complete RPMI-1640 medium and counted by Trypan Blue exclusion method. All experiments with spleen cells were carried out in complete R10 unless otherwise indicated.
Infection of splenocytes with BCGGFP
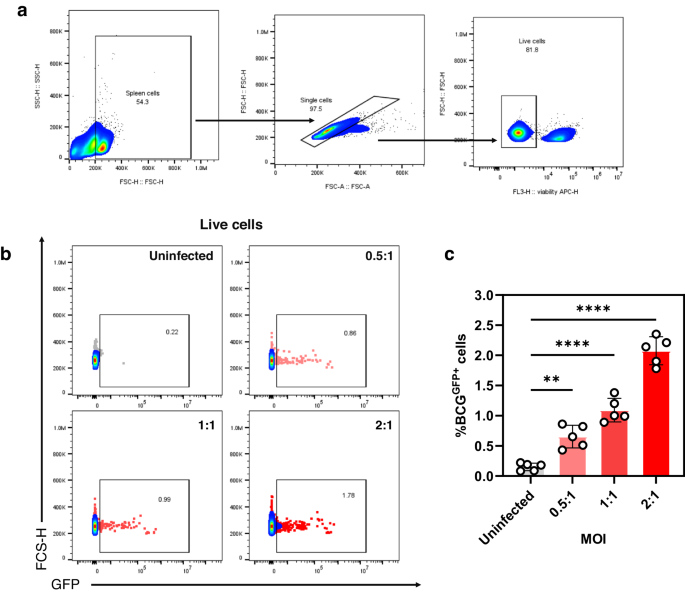
One million spleen cells were seeded in 96 flat-bottom well plates. MOIs were either 0.5, 1, or 2. BCGGFP infection of splenocytes was carried out for 24 h. Wells were washed thrice with 200 µL sterile DPBS prior to FACS staining and analysis.
Conventional and modified mycobacterial growth inhibition assay
A modMGIA was used to assess the capacity of mouse splenocytes for in vitro killing of BCG or Mtb (Supplementary Fig. 1). Initial experiments using BALB/c mice and two congenic cell lines (MH-S and J774) were done to test different conditions including cell density in different well plates (96- and 48-well plates), and ratio of splenocytes to cells infected with mycobacteria. For these experiments, cell lines were infected as above mentioned (MOI 0.1 or 1:1) for 48 h before coculture, culture medium was removed and then replenished with fresh medium (100 µL for 96-well plate and 700 µL for 48 well plates). This was followed by the addition of splenocytes to designated wells (100 µL of 1 × 106 for 96-well plates and 300 µL of 3 × 106 for 48 well plates). For conventional MGIA, 3 × 106 splenocytes plated in 48 well plates were directly infected with 500 CFUs of Mtb. Both conventional and modified MGIA cultures were left for 120 h, followed by aspiration of the media, and cell lysis with sterile distilled water. Cell lysates were used for CFU- or luciferase-based assays to quantify viable mycobacteria. In some experiments, aspirated media were collected for secreted cytokine analysis.
Cell surface staining and flow cytometry analysis
Splenocytes or cell lines were added to 96-well U-bottom plates and centrifuged at 300×g for 5 min. Cells were then washed with sterile DPBS thrice, then stained using a cocktail of antibodies (1:200 PE anti-mouse H-2, Clone M1/42, BioLegend® 125506; and 1:200 Brilliant Violet 510TM anti-mouse I-A/I-E, Clone M5/114.15.2, BioLegend® 107635), 1:500 eBioscienceTM fixable viability dye eFluorTM 780 (Invitrogen,65-0865-14), and 1:250 mouse Fc block (TruStain fcX™ [anti-mouse CD16/32]) (BioLegend®, 422302) for 30 min at 4 °C. Afterwards, cells were washed three times then resuspended in sterile 1X PBS prior to FACS acquisition using CytoFlex (Beckman Coulter). For all experiments, at least 20,000 live events were obtained per sample. Flow cytometry results were analysed using FlowJoTM v.10.8 (BD Life Sciences). Representative gating strategy and histograms are shown in Supplementary Fig. 2.
Cytokine ELISA using cell culture supernatants
Commercially available kits (all from Invitrogen) were used to quantify IFNγ (88-7314-88) and TNFα (88-7324-88) from culture supernatants. Plates were coated with 100 µL capture antibody in PBS overnight at 4 °C. After three wash steps with PBS + 0.05% Tween-20 (PBS-T), wells were blocked with 200 µL blocking buffer for 1 h at room temperature. Wells were then washed three times, then 100 µL of neat supernatants or serial dilutions of the standards were added to wells. After 2 h of incubation at room temperature, wells were washed five times with PBS-T, followed by the addition of 100 µL of detection antibody. Plates were incubated at room temperature for an hour and washed five times with PBS-T. Wells were then incubated for 1 hr at room temperature with 100 µL of streptavidin-HRP. After seven washes with PBS-T, TMB substrate was added to each well. Plates were left to develop for 15 mins, followed by the addition of stop solution (0.16 M H2SO4, In-house). The absorbance reading for each plate was obtained at 450 nm with a correction of 540 nm using a plate reader (Tecan, UK). Absorbance readings from blank wells were subtracted from all wells with samples or standard.
Confocal microscopy
To visualise internalisation of BCGGFP by cells, 50,000 cells were added to 96-well clear bottom black plates (Corning Inc., 3603). After 2 h, BCGGFP was added (MOI = 4) to cells followed by incubation for 24 h. Wells were gently washed with DPBS thrice, followed by cold fixation and permeabilization for 15 min using IC fixation buffer (Invitrogen, 00-822-49). Wells were then gently washed twice with 1X permeabilization buffer (Invitrogen, 00-8333-56), followed by staining for F-actin using Phalloidin-iFluor647 (1:2000) (Abcam, ab176759) for 45 min in 4 °C. After three washes with 1X permeabilization buffer, nuclear counterstaining for 5 min was done using 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (1:1000) (Thermo Fisher Scientific, D1306). Stained cells were finally resuspended in DPBS, followed by image acquisition using Nikon A1R confocal microscope and NIS Elements software. Images were generated using NIS Viewer v 5.21.00 (Nikon, UK).
Statistical analysis
All statistical analyses were performed in GraphPad Prism Version 9.2.0 (La Jolla, CA). Where applicable, unpaired t-test, one-way ANOVA followed by Dunnett’s or Tukey’s test for multiple comparisons, or two-way ANOVA followed by Tukey’s range test as post-hoc analysis was done. All data shown are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 were considered significant. Intra-assay and inter-assay coefficient of variations (CV) were calculated using the formula: (standard deviation/mean × 100). Intra-assay precision represents the means between CV of PBS and BCG groups, and inter-assay precision is the mean CV of results from modMGIA.


留言 (0)