Patients and clinical prostate specimens
Tumor and adjacent tissues from 38 PCa patients were collected from January 2020 to December 2020. All patients were first diagnosed with PCa at Shengjing Hospital of China Medical University and did not have other malignancies. The study was conducted following the Declaration of Helsinki (as revised in 2013), and written consent for tissue donation was obtained from each patient. The protocol was approved by the Institutional Review Board of Shengjing Hospital of China Medical University (approval no. 2019PS1154K; approved date 10 June 2019).
Cell culture, cell transfection, and establishment of stable cell lines
Human prostate epithelial cells (HPECs, cat. no. CP-H019) and PCa cell lines LNCaP (cat. no. CL-0143) and PC-3 (cat. no. CL-0185) were purchased from Procell (Wuhan, Hubei, China). All cells were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C with 5% CO2.
Overexpression lentivirus (oeFAM84B, oeWWP1, oeCDKN1B) based on mammalian gene expression lentiviral vector (pLV[Exp]-EGFP:T2A:Puro-EF1A), shLig3 1, 2, 3#, shWWP1 1, 2, 3# based on mammalian short hairpin (sh)RNA interference lentiviral vector (pLV[shRNA]-EGFP:T2A:Puro-U6), and their respective controls (oeCtrl and shCtrl) were purchased from VectorBuilder (Guangzhou, Guangdong, China). The virus titer was 109 TU/mL, and the corresponding lentivirus was used to infect PCa cells (multiplicity of infection = 5) for 48 h. Stably transfected cell lines were subsequently screened with 2 μg/mL puromycin for 2 weeks. ShRNAs sequences specific for Lig3 and WWP1 were: shLig3 1#: CCGGATCATGTTCTCAGAAATCTCGAGATTTCTGAGAACATGATCCGG; shLig3 2#: GCCCACTTTAAGGACTACATTCTCGAGAATGTAGTCCTTAAAGTGGGC; shLig3 3#: CAGGAGTCATTAAGACTGTTTCTCGAGAAACAGTCTTAATGACTCCTG; shWWP1 1#: GACTTGAGGAGGCGCTTATATCTCGAGATATAAGCGCCTCCTCAAGTC; shWWP1 2#: CATGGAATCTGTCCGAAATTTCTCGAGAAATTTCGGACAGATTCCATG; shWWP1 3#: GCTGTTCAGAAAGGTATTAAGCTCGAGCTTAATACCTTTCTGAACAGC.
Stably transfected PCa cells were treated with MYC inhibitor MYCi361 (S8905, Selleck, Houston, TX, USA) at 6 μM for 3 h to promote MYC protein degradation [14] or Wnt signaling pathway inhibitor LF3 (S8474, Selleck) at 30 μM for 4 h to disrupt the interaction between beta catenin and TCF4. Dimethylsulfoxide (DMSO) was used as the control for both treatments.
Outward and inward PCR
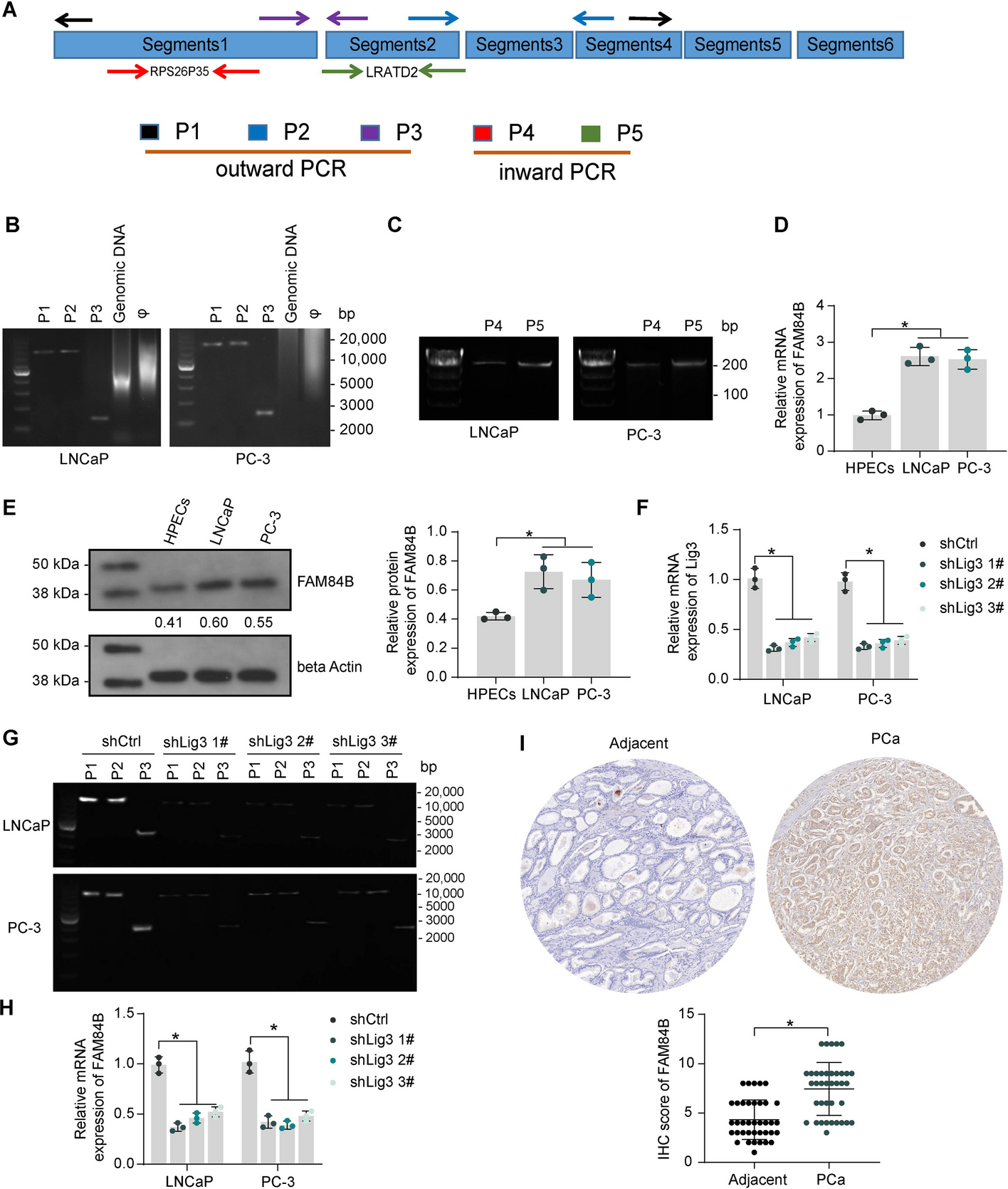
To validate the circular structure of eccDNA 3#, outward PCR primers containing different junction sites of eccDNA 3# and inward PCR primers (Table 1) targeting representative intact transcripts in different segments were designed. All reactions were performed using human genomic DNA as a control, and the PCR reaction system contained a phi29 amplification template, primers, and master mix for PCR (#1665009EDU, Bio-Rad Laboratories, Hercules, CA, USA). PCR assays were performed in a PCR cycler under standard PCR conditions according to the manufacturer’s protocols, and the circular structure of eccDNA 3# was confirmed based on agarose gel electrophoresis PCR.
Table 1 Primer for outward and inward PCRWestern blot
Total proteins were extracted from the cells using RIPA lysis buffer (Roche Diagnostics, Co., Ltd., Rotkreuz, Switzerland) containing protease and phosphatase inhibitors. Equal amounts of protein samples were separated by 8–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to PVDF membranes (Millipore Corp, Billerica, MA, USA). Primary antibodies were incubated overnight at 4 ℃, followed by reprobing with the secondary antibody. Signals were subsequently enhanced with chemiluminescent reagents (Abcam, Cambridge, UK). Primary antibodies used were FAM84B (1:1000, 18421-1-AP, ProteinTech Group, Chicago, IL, USA), MYC (1:1000, #18583, Cell Signaling Technologies, Beverly, MA, USA), beta catenin (1:2000, 17565-1-AP, Cell Signaling Technologies), WWP1 (1:2000, 28689-1-AP, ProteinTech), CDKN1B (1:5000, ab32034, Abcam), beta actin (1:200, ab115777, Abcam), and ubiquitin (1:1000, #20326, Cell Signaling Technologies).
PCR analysis
Total RNA was extracted from the cells using TRIzol reagent (GK20008, Glpbio, Montclair, CA, USA). After reverse transcription with the iScript cDNA Synthesis kit (#1708890, Bio-Rad Laboratories), the SsoAdvanced Universal SYBR Green Supermix (#1725270, Bio-Rad Laboratories) was used for qPCR reactions on a CFX Opus 96 Real-Time PCR system (Bio-Rad Laboratories). The relative mRNA levels were normalized using beta actin as a control. The 2−∆∆Ct method was used to analyze the results. The primer sequences are presented in Table 2.
Table 2 Primers used for qPCRImmunohistochemistry (IHC)
Paraffin-embedded tissue sections (4 μm) were dewaxed using xylene and dehydrated with gradient ethanol, followed by the addition of 3% hydrogen peroxide to block endogenous peroxidase activity. Antigen retrieval was performed by heating sections in sodium citrate buffer (pH 6.0) in a microwave oven at 100 °C for 30 min, followed by detection of immunoreactivity by the Rabbit-diaminobenzidine Detection IHC kit (IHC0007, FineTest, Wuhan, Hubei, China). Briefly, nonspecific antigen binding was blocked by blocking serum, and then the sections were stained with primary antibody overnight at 4 °C, followed by color development by DAB after incubation with poly-horseradish peroxidase-Goat Anti-Rabbit IgG at room temperature for 1 h. Hematoxylin was used to counter-stain the nuclei. Scoring was performed for clinical samples and was completed by three pathologists who were unaware of the grouping. The score was determined according to the positive staining intensity (0: negative; 1: weak; 2: moderate; 3: strong) and positive staining cells quantity: (0: < 5%; 1: 5~25%; 2: 25~50%; 3: 50~75%; 4: > 75%), and the final score was intensity score × quantity score (0–12). Xenograft tumor tissues were viewed by microscopy, and positively stained areas were quantified using Image J software. Antibodies to FAM84B (1:200, 18421-1-AP, ProteinTech), Ki67 (1:200, 28074-1-AP, ProteinTech), WWP1 (1:500, 28689-1-AP, ProteinTech), Lig3 (1:200, A22136, ABclonal, Wuhan, Hubei, China), MYC (1:200, #18583, Cell Signaling Technologies), and CDKN1B (1:50, ab32034, Abcam) were used.
Proliferation and viability assays
The treated PCa cells were suspended with 100 μL of the medium, seeded into 96-well plates (3000 cells/well), and incubated for the indicated periods (1, 3, 5 days). After the addition of 10 μL of Cell Counting Kit-8 (CCK8; GK10001, Glpbio) to each well, the incubation continued for 2 h in a cell incubator. The cell proliferation was measured by reading the OD value at 450 nm using a microplate reader.
The DNA synthesis of the cells was determined using the BeyoClick EdU-594 Cell Proliferation Assay kit (C0078S, Beyotime, Shanghai, China) according to the supplier’s protocol. Treated PCa cells (1 × 104) were placed in a 96-well plate and treated with 50 μM of EdU for 2 h. Afterward, the cells were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature and permeabilized with 0.5% TritonX-100 for 10 min, followed by staining with Click Additive Solution for 30 min in the dark. Hoechst 33342 was used to stain cells for 5 min in the dark to label the nucleus. Finally, the cells were observed by fluorescence microscopy (Olympus, Tokyo, Japan), and the proportion of EdU-positive cells was calculated.
Migration and invasion assays
Transwell chambers (8-μm pore size, Corning Costar, Corning, NY, USA) were used for cell migration and invasion assays. For the migration assay, 1 × 105 treated PCa cells were seeded in the apical chamber containing serum-free medium, and a complete medium containing 10% FBS was supplemented to the basolateral chamber. After 24 h, migrated cells were fixed with 4% paraformaldehyde and stained with crystal violet. For the invasion assay, the apical chamber was precoated with Matrigel, and the rest of the steps were the same as for the migration assay.
Chromatin immunoprecipitation (ChIP) assay
The promoter sequence of MYC (chr8:127736036–127736381) and the WWP1 promoter sequence (chr8:86341669–86343512) with a potential binding relationship to MYC were obtained from the UCSC Genome Browser (https://genome.ucsc.edu/index.html). The SimpleChIP Plus Enzymatic ChIP kit (#9005, Cell Signaling Technologies) was used to detect the ability of MYC to recruit the WWP1 promoter and and the ability of beta catenin to recruit the MYC promoter. The cells were fixed with formaldehyde and then lysed, and chromatin was partially digested with micrococcal nuclease to form fragments. The chromatin fractions were incubated with antibodies to one of the following: MYC (1:100, #18583, Cell Signaling Technologies), beta catenin (1:100, 17565-1-AP, Cell Signaling Technologies), or normal rabbit IgG with ChIP-grade protein G magnetic beads. After protein–DNA was de-crosslinked, purification was performed using DNA purification centrifuge columns, and enrichment of the WWP1 promoter or MYC promoter was detected by qPCR reaction. The qPCR reaction system consisted of nuclease-free H2O, 5 µM promoter primer, and SimpleChIP universal qPCR premix. A total of 40 cycles of the standard PCR reaction program were performed according to the manufacturer’s protocol. The signal obtained from each immunoprecipitation was expressed as a percentage of the total input chromatin: % of input = 1% × 2(Ct 1%input sample−Ct IP sample), Ct = Threshold cycle of PCR reaction.
Luciferase reporter assay
The WWP1 promoter sequence (chr8:86341669–86343512), which has a binding relationship with MYC or the MYC promoter sequence (chr8:127736036–127736381) was inserted into the pGL3 basic vector (Promega Corporation, Madison, WI, USA) to construct the WWP1 or MYC promoter luciferase reporter plasmid. These plasmids were transfected into treated PCa cells by Lipofectamine 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA), and the luciferase activity was assessed by a dual luciferase reporter assay system (Promega) after 48 h.
TOP/FOP flash
TOP flash plasmid (D2501, Beyotime) containing the TCF/LEF binding site was used to detect Wnt/β-catenin pathway activity in cells, and the FOP flash plasmid (D2503, Beyotime) containing the mutated TCF/LEF binding site sequence was used as a negative control. The above plasmids were transfected into the treated PCa cells by Lipofectamine 2000 (Thermo Fisher), and the luciferase activity was measured by Dual-Luciferase Reporter Analysis System (Promega Corporation, Madison, WI, USA) after 48 h.
Formation of xenografts and metastases
The protocols were approved by the Animal Research Ethics Board of Shengjing Hospital of China Medical University (approval no. 2022PS1170K; approved date 4 January 2022) following the Guidelines for the Care and Use of Animals. The study involving animals was conducted following the Basel Declaration. Seven-week-old male NOD/SCID mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and acclimatized for 1 week before the experiment. All mice were randomly divided into 12 groups of 10 mice each.
Five mice in each group were randomly selected for tumor growth experiments. The PC-3 cells were suspended in PBS at a density of 1 × 107 cells/mL, and Matrigel (Corning) was added to the cell suspension at a ratio of 1:1. The mixture (150 μL) was injected subcutaneously into the back of mice to induce tumor growth. The tumor volume (V) was measured once a week with calipers, and the formula is as follows: V = 0.5 × L × W2, where L = length and W = width. After 4 weeks, the mice were euthanized, and their tumor size and weight were measured.
The remaining five mice in each group were used for tumor metastasis experiments. PC-3 cells were labeled with luciferase and treated as indicated. Subsequently, 100 µL (1 × 107 cells/mL) of the cell suspension was injected intracardially into the left ventricle. Each week, 150 µL of 30 mg/mL d-luciferin was injected intraperitoneally into each mouse, and then luciferase activity was measured using the IVIS Small Animal Live Imaging System to assess tumor metastasis. For mice requiring MYCi361 treatment, MYCi361 (55 mg/kg/day) was administered by gavage after tumor cell injection for 3 consecutive days per week for 2 weeks.
Co-immunoprecipitation (Co-IP)
PCa cells in the oeCtrl and oeWWP1 groups were pretreated with MG-132 (S2619, Selleck) at a concentration of 100 nM for 12 h to inhibit 26S proteasome-mediated protein degradation, followed by lysis in RIPA lysis buffer and centrifugation. A portion of the supernatant was used as input, and the other supernatant was immunoprecipitated with CDKN1B antibody (1:30, ab32034, Abcam) overnight at 4 °C and incubated with protein A/G agarose beads (Thermo Fisher Scientific) for 60 min at room temperature. The immune complexes were washed with RIPA buffer containing 5% Tween-80 and assayed by western blot assays.
Determination of protein stability
Cycloheximide (CHX, S7418, Selleck) was used to treat PCa cells in the oeCtrl and oeWWP1 groups at a concentration of 500 nM for 0, 3, and 6 h to inhibit protein synthesis. The cells were collected at each time point, and CDKN1B protein expression was detected by western blot assays to assess the protein stability of CDKN1B.
Data analysis
Experiments were carried out at least three times unless otherwise indicated. Statistical analysis was performed using GraphPad Prism 8.0.2 software (GraphPad, San Diego, CA, USA). All data were described as mean ± standard deviation (SD) if applicable. Statistical difference was performed with paired t-test (two groups) or one-way/two-way analysis of variance (ANOVA) (three or more groups), followed by Tukey’s or Dunnett’s post hoc test, where appropriate. For the correlation between genes, data were compared using Pearson’s correlation analysis, and for the correlation between gene expression in PCa tissues and clinical parameters in patients, a Chi-square test was conducted. p-value < 0.05 was considered statistically significant.





留言 (0)