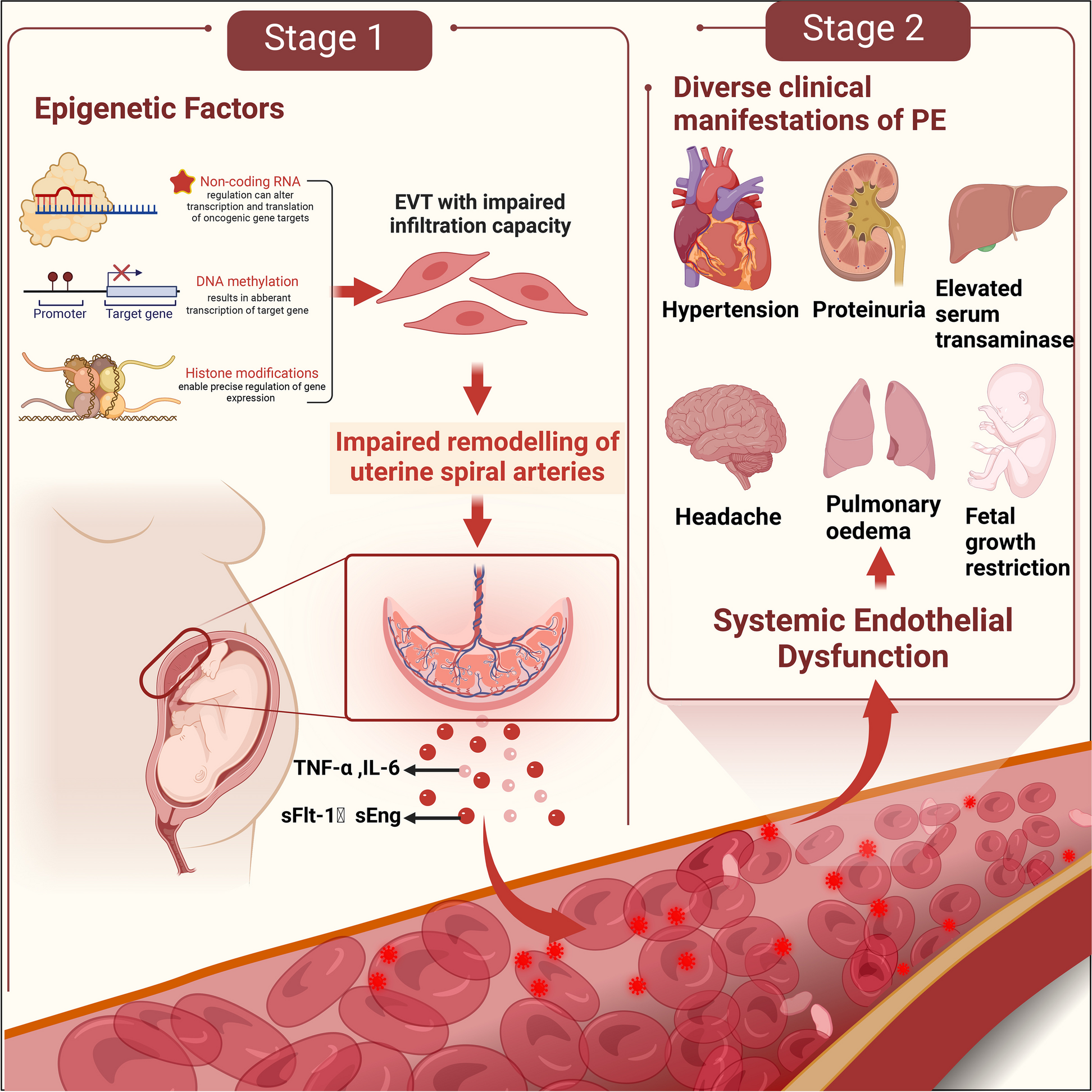
Animals and treatment
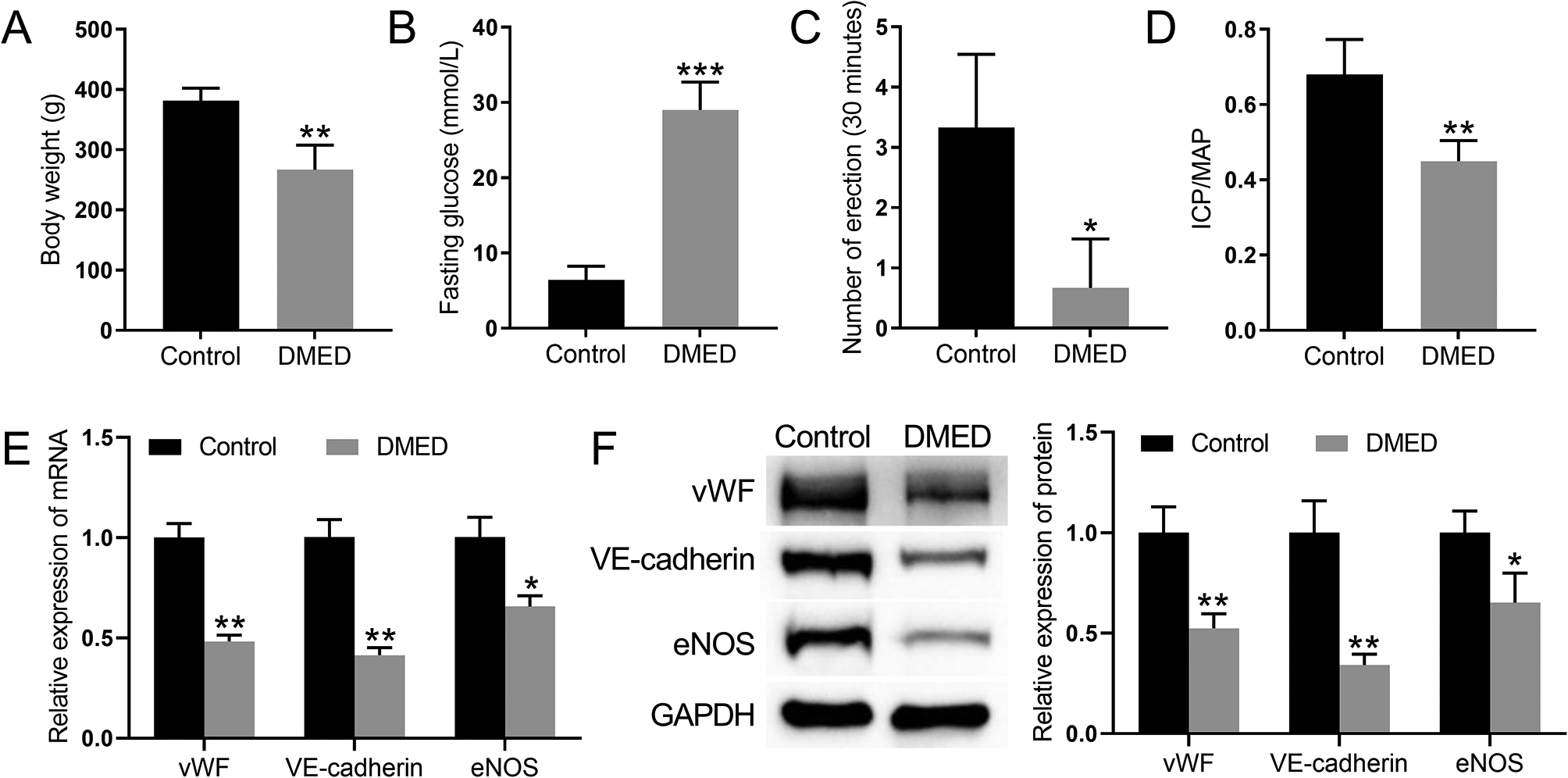
Male Sprague–Dawley (SD) rats weighing 250–300 g (10 weeks old) were purchased from Beijing Weitong Lihua Laboratory Animal Science and Technology Co., Ltd., and a mating test confirmed that they had normal erectile function. The rats were randomly divided into experimental and control groups after 1 week of free-feeding and water-acclimatization. Rats were injected intraperitoneally with 1% streptozotocin (Solarbio, Beijing, China) to construct a DM rat model, and blood glucose levels were measured after 72 h. Rats with blood glucose levels higher than 16.7 mmol/L were considered successful DM models. Diabetic rats were fed for 8 weeks to develop ED. Rats without penile erection were considered DMED rats, and erectile function was assessed using the apomorphine (APO)-induced erection test (Solarbio, Beijing) [24]. After the DMED model was established in the experimental group of rats, experiments were conducted by implanting PBS, BM-MSCs, BM-MSCs overexpressing MALAT1, or BM-MSCs combined with sildenafil (Solarbio, Beijing). The animal experiments were approved by the Ethics Committee of The First Affiliated Hospital of Nanchang University (CDYFY-IACUC-202312QR023).
Intracavernosa pressure (ICP) and mean arterial pressure (MAP) assessment
ICP and MAP assessments were performed as previously described [10]. In brief, 3% sodium pentobarbital solution was injected intraperitoneally to anesthetize the rats. The rats were immobilized, and the abdomen was disinfected with iodophor. Two PE50 tubes connected to a pressure-sensing device were placed into the left common carotid artery and the cavernous body of the penis for pressure measurement. The cavernous nerves located in the dorsal aspect of the rat prostate were isolated using a microscopic instrument. Electrical stimulation was performed with a bipolar hook 3–5 mm from the pelvic ganglion for 60 s, resulting in different degrees of penile erection. The physiological pressure signal was transduced by a physiological signal transducer, and the ICP curve was recorded.
Cell culture and treatment
The rats were sacrificed by cervical dislocation. After isolating the femur, and the bone marrow cavity was exposed and repeatedly washed with low-glucose DMEM (Gibco, Grand Island, NY, USA). The medium containing bone marrow was centrifuged at 1,000 r/min for 10 min, and the supernatant was discarded. The obtained cells were cultured at 37 °C in an incubator containing 5% CO2. Cell growth was observed by inverted microscopy. BM-MSCwere treated with 20, 30, 40, or 50 ng/ml VEGFA to induce differentiation towards endothelial cells [11].
Cell transfection
The cells were transfected as previously described [25]. In brief, MALAT1 or CDC42 cDNA was cloned and inserted into the pcDNA3.1 vector (Invitrogen, Thermo Fisher Scientific). An empty pcDNA3.1 vector was used as a negative control (NC). GenePharma (Shanghai, China) provided the miR-206 mimic/inhibitor and the corresponding control mimic/inhibitor NC, RNA oligonucleotides for MALAT1 knockdown (si-MALAT1), and RNA oligonucleotides for CDC42 knockdown (si-CDC42). Subsequently, the BM-MSCs were transfected using Lipofectamine® 3000 (Invitrogen, USA) before chemical treatment.
qRT‒PCR
Total RNA from rat cavernosum tissue was extracted by adding TRIzol (Thermo Fisher Scientific), chloroform, and isopropanol according to the manufacturer’s instructions. DNA was removed from the extracted total RNA with a DNA Eraser Buffer kit (TaKaRa, Dalian, China). The obtained RNA was reverse transcribed using a PrimeScript RT Enzyme Mix I kit (TaKaRa). After the reaction was completed, primers were added for PCR amplification. The thermocycler reaction conditions were as follows: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, 55 °C for 30 s, and 72 °C for 30 s. The final results were analysed by the 2−ΔΔCT method.
Western blot analysis
BM-MSCs in the logarithmic growth phase were washed twice with PBS (Gibco, Grand Island, NY, USA) and lysed with RIPA buffer (Beyotime, Shanghai, China). Total protein (30 µg) was utilized for Western blot analysis. The membranes were incubated overnight at 4 °C with the following primary antibodies: anti-vWF (1:200), anti-VE-cadherin (1:500), anti-eNOS (1:500), anti-MALAT1 (1:500), anti-CDC42 (1:500), anti-PAK1 (1:500), anti-paxillin (1:500), anti-PY31 (1:500), and anti-PY118 (1:500). The membranes were then incubated at room temperature for 1 h with a goat anti-rabbit IgG secondary antibody (1:3,000), and the target bands were visualized and analysed.
Total protein was extracted from the penile cavernosum and lysed with RIPA buffer after grinding in a low-temperature homogenizer, and 60 µg of each sample was utilized for Western blot analysis. The membranes were incubated at 4 °C overnight with the following primary antibodies: anti-vWF (1:200), anti-VE-cadherin (1:500), anti-eNOS (1:500), anti-CDC42 (1:500), anti-PAK1 (1:500), anti-paxillin (1:500), anti-PY31 (1:300), and anti-PY118 (1:500). The membrane was then incubated with a goat anti-mouse IgG secondary antibody (1:3000) at room temperature for 1 h, and the bands were analysed with ImageJ software.
Coimmunoprecipitation (Co-IP) assay
Coimmunoprecipitation was performed as previously described [26]. Briefly, the collected cells were washed three times with 1× PBS and then lysed on ice in IP lysis buffer containing protease inhibitor cocktail for 1 h. The protein concentration was determined by the BCA method. After preparing the system, 40 L of protein A + G microspheres was added to the extracted protein, followed by incubation at 4 °C for 1 h. The samples were then centrifuged at 14,000×g for 2 min, and the supernatants were removed. Then, 10 L of polyclonal CDC42, PAK1, or paxillin antibody was added, followed by incubation at 4 °C overnight. The samples were centrifuged at 10,000×g for 20 s, and the supernatants were removed. After collecting the microspheres, the samples were washed with PBS, and 50 L of SDS Sampling Buffer was added. The mixture was heated to 85 °C for 10 min and centrifuged at 5,000×g for 10 min. The supernatant was then collected for protein electrophoresis.
Transwell migration assay
After trypsin digestion, the cell concentration was adjusted to 5 × 104 cells/mL in serum-free DMEM. For the Transwell assay, 200 µL of cell suspension was added to the upper chamber, and DMEM containing10% FBS was added to the lower chamber. The chamber was removed after 24 h, and the cells at the bottom of the upper chamber were removed with a cotton swab. The cells were fixed with 4% paraformaldehyde for 15 min, washed with PBS, stained with 0.5% crystal violet for 15 min. After washing, the cells were imaged under a microscope, and five fields of view were randomly selected for counting. The results of three replicate experiments were analysed statistically analysed.
Luciferase assay
Wild-type and mutant MALAT1 (with a mutated miR-206 binding site) were cloned and inserted into the pmirGLO dual luciferase vector (GenePharma). BM-MSCs were cotransfected with wild-type pmirGLO-MALAT1 (or mutant) and miR-206 mimics (or negative controls) using Lipofectamine 2000. Similarly, dual-luciferase reporter plasmids containing CDC42-WT and CDC42-MUT were constructed. BM-MSCs were cotransfected with miR-206 mimics and luciferase reporter plasmids or their corresponding empty vector using Lipofectamine 2000. At 48 h after transfection, luciferase activity was analysed using a dual-luciferase reporter kit (Promega, USA).
Immunohistochemistry
The 10% formaldehyde-fixed tissues were dehydrated by an automatic dehydrator, embedded, and sectioned. The sections were deparaffinized, immersed in 3% methanol hydrogen peroxide for 10 min, and washed three times with PBS (5 min each wash). The sections were heated to boiling in citrate buffer, washed with PBS, and immersed in blocking solution for 20 min. Primary CDC42 and PAK1 were then added and incubated overnight. Biotinylated secondary antibody was added dropwise, and the sections were incubated at 37°C for 30 min. After wash three times with PBS, the sections were stained with 3,3’-diaminobenzidine (DAB). The sections were sealed with clear gum, and photomicrographs were acquired under a light microscope. The results were analysed using Image-Pro Plus 6.0.
Statistical analysis
Each assay was performed three times. The data were analysed by SPSS 22.0 statistical software (IBM, Armonk, NY, USA) and are expressed as the mean ± standard deviation. Independent sample t tests and one-way ANOVA were used to detect differential expression of the indicators. P < 0.05 was considered to indicate statistical significance.





留言 (0)