Experimental Animals
In this study, male mice aged 7–8 weeks and weighing–23–26 g were used. These mice were obtained from the animal house facility at the Hashemite University. They were kept under controlled conditions in the animal house, with regular access to food and water and a 12-h light-dark cycle. Before starting the experiments, mice were allowed to adapt to the laboratory environment for at least 2 h. Each mouse was used only once in this study. The study adhered to the Ethics Committee of Hashemite University's approval (IRB number: 14/4/2021/2022, dated April 14, 2022) and followed the ARRIVE guidelines for ethical animal research reporting.
Colitis Induction and Study Design
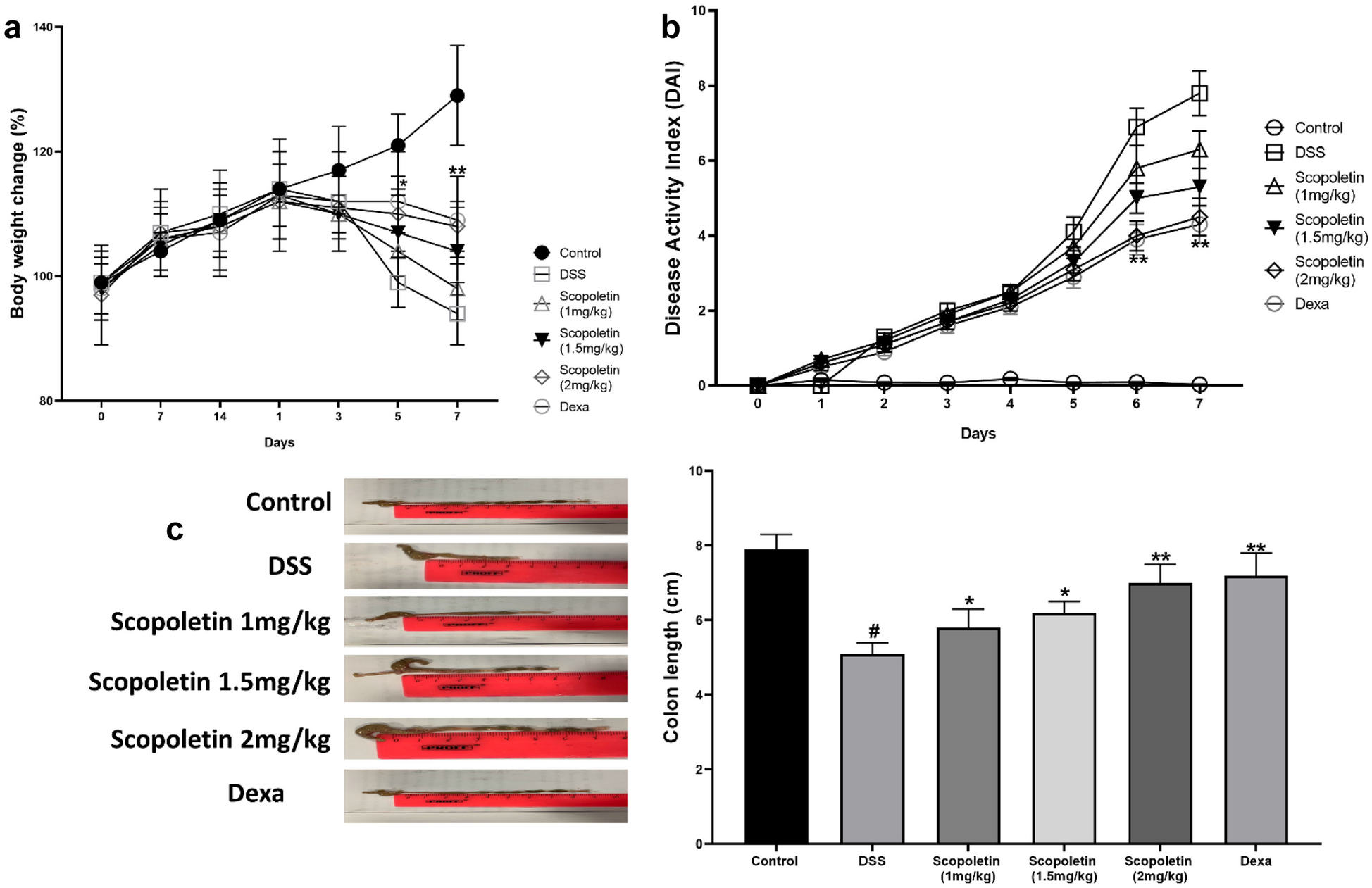
To induce acute colitis, mice were administered 2.5% (w/v) DSS (Sigma-Aldrich, Gillingham, Dorset, UK) in drinking water for 7 days. The mice were divided into six groups of ten mice each. The control group received water only. DSS group was given DSS. Scopoletin-treated groups with different doses of (1 mg/kg, 1.5 mg/kg, and 2 mg/kg, respectively) along with DSS.Dexamethasone (2 mg/kg) was orally administered to the final group as a standard reference drug. The doses of scopoletin (1, 1.5, and 2 mg/kg) were chosen based on the preliminary data (Supplementary Fig. 1). A pilot experiment conducted prior to the main study assessed the dises activity index (DAI) across a range of doses of scopoletin in a subacute model of colitis. These preliminary investigations revealed that doses below 1 mg/kg were less effective in ameliorating colitis symptoms, whereas doses exceeding 2 mg/kg did not provide additional therapeutic benefits, and were associated with an increased risk of toxicity. Therefore, the selected doses were deemed to offer an optimal balance between efficacy and safety.
Socopoletin was administered orally to Groups III to V, starting 14 days prior to and during the DSS treatment, once daily. Group VI was administered with socopoletin (2 mg/kg) throughout the experiment. The control and DSS groups were administered with water instead of socopoletin.
The weights of the mice were recorded on specific days (0, 7, 14, 15, 16, 17, 18, 19, 20, and 21) by using an electronic analytical balance (Sartorius Products, Germany). The Disease Activity Index (DAI) was evaluated as described by Alex et al. [24]. Briefly, a comprehensive scoring system was employed to quantitatively assess the key clinical parameters of disease activity, including body weight loss, stool consistency, rectal bleeding, and the overall clinical condition of the mice. Each parameter was assigned a score, as follows:
Body Weight Loss: Scores were assigned based on the percentage of weight loss compared to baseline: 0% (score 0), 1–5% (score 1), 5–10% (score 2), 10–15% (score 3), and > 15% (score 4).
Stool Consistency: Normal stools were scored as 0, soft but formed stools as 1, very soft stools as 2, and liquid stools as 3.
Rectal Bleeding: No scored as 0, hemoccult positive as 1, visible blood traces in the stool as 2, and gross bleeding, 3.
Overall Clinical Condition: Mice were observed for signs of distress, including activity level and posture, with scores ranging from 0 (normal behavior) to 3 (severe distress).
The cumulative Disease Activity Index (DAI) was calculated by summing the scores of these parameters, providing a comprehensive assessment of colitis severity ranging from 0 (no disease) to 13 (maximum disease severity).
After 21 days, the mice were euthanized by cervical dislocation under anesthesia. Their colons, from the cecum to 1 cm above the anus, were measured and prepared for histological examination with hematoxylin and eosin (H&E) staining using a previous scoring system.
Myeloperoxidase (MPO) Assay
MPO assay was utilized to evaluate MPO activity, serving as an indicator of neutrophil presence and distribution in tissue samples. Initially, the colonic tissues were weighed and homogenized in phosphate-buffered saline (PBS) at a predetermined ratio of 1:9, based on the tissue weight to PBS volume.
After homogenization, the mixture was centrifuged to separate the supernatant containing MPO. Supernatants were collected for further analysis. The measurement of MPO activity proceeded according to the prescribed procedures of the MPO assay kit's manufacturer (Abcam, Cambridge, UK). Adhering to these standardized instructions is vital for the consistent and precise evaluation of MPO activity, thereby providing an accurate gauge of neutrophil infiltration levels in the colonic tissues.
Intestinal Bacteria Cultivation and Quantitation
The objective of the experiment was to quantify the specific intestinal bacteria on the final day (day 21). Fecal pellets were collected, weighed, and homogenized in 1 ml of sterile PBS. This homogenate was then subjected to a sequence of serial dilutions that were carefully plated onto distinct media, each tailored to different bacterial types. These included MRS medium for lactobacilli, BSM Medium for bifidobacteria, and MacConkey Agar for gram-negative bacteria, all sourced from Sigma-Aldrich (Gillingham, Dorset, UK).
Plated cultures were then incubated under meticulously regulated conditions suitable for growth. Cultures on MRS and BSM, aimed at cultivating lactobacilli and bifidobacteria, respectively, were incubated anaerobically for 2–3 days at 37 °C. Conversely, MacConkey Agar plates, designed for gram-negative bacteria, underwent aerobic incubation, but only overnight at 37 °C incubation, colonies displaying the characteristic features of their respective bacterial groups were counted. For a valid and comprehensive count of the bacterial colonies, only plates with colony counts ranging from 30 to 300 were considered. This specific range was chosen to circumvent potential errors, such as overcounting in densely populated plates or undercounting in sparsely populated plates. Thus, this approach ensured a reliable measure of bacterial populations in fecal matter.
Preparation of Caecal Bacterial Lysates
The Caecal Bacterial Lysates (CBL) were prepared according to the methodology described by Dieleman et al. [25]. This commenced with the extraction of cecal content from the mice, which was then thoroughly mixed in RPMI 1640 medium through vigorous vortexing. This step ensured a homogeneous suspension of cecal material in the medium. Subsequently, the mixture was incubated with 10 μg/ml DNase and 0.01 M MgCl2. The inclusion of DNase is crucial for degrading any DNA present, whereas MgCl2 serves as a catalyst for enhancing the enzymatic function of DNase incubation, the sample underwent for 3-min homogenization using 0.1 mm glass beads in a Mini-Bead Beater. This method, which utilizes glass beads, is a standard method for physically breaking bacterial cell walls, thereby releasing cellular content into a homogenate. The homogenate was then centrifuged at 10,000 × g for 10 min, a critical step for separating bacterial cell debris from the supernatant containing bacterial lysates.
The final stage involved refining the supernatant by passing it through a 0.45 μM syringe filter. This filtration process effectively removed any residual particulate matter and yielded a clear lysate. The resulting filtered CBL was set aside for subsequent experimental use or analysis as required by the study.
Mesenteric Lymph Node Cell Cultures
Mesenteric lymph nodes (MLN) from mice were meticulously harvested following the method outlined by Ruyssers et al. [26]. The initial phase of this process involves the creation of single-cell suspensions from the lymph nodes. This step is critical because it ensures that the cells are individually separated, thereby facilitating their independent analysis or utilization in further experiments.
Once the single-cell suspensions were established, approximately 4 × 105^5 MLN cells were extracted and mixed with 20 μg/ml of Caecal Bacterial Lysates (CBL). The mixture was then placed in 96-well flat-bottom microplates. The microplates were filled with RPMI 1640 medium supplemented with 5% heat-inactivated fetal calf serum and 50 mg/ml gentamicin. Fetal calf serum provides vital nutrients and growth factors necessary for cell sustenance, while gentamicin helps prevent bacterial contamination within the culture.
Subsequently, these cultures were incubated in a carefully controlled environment that was optimal for cell culture. This environment was maintained at 37 °C in a 5% CO2 atmosphere, closely simulating natural bodily conditions, to foster cell growth and survival. The incubation duration was set at 72 h, allowing ample time for the cells to interact with CBL and trigger potential cellular responses.
Following the incubation period, the next critical stage involved the collection of culture supernatants. These supernatants are significant because they contain cytokines secreted by MLN cells in reaction to CBL. Cytokines play a vital role as indicators of immune responses, making them a primary focus in experimental settings. The collected supernatants were preserved at -20 °C for later analysis. Storing the samples at this temperature is crucial for maintaining the integrity of cytokines and other components in the supernatants, ensuring their stability for precise and accurate cytokine measurements in subsequent evaluations.
Cytokine Assays
For cytokine analysis, the colon samples were first weighed and then meticulously blended with phosphate-buffered saline (PBS) at a ratio of 1 part sample to 9 parts PBS while being maintained on ice. This step ensured the proper dilution and preservation of the samples for analysis. Subsequently, the blended mixture was centrifuged at 2, 000 × g for 40 min at a controlled temperature of 4 °C. This centrifugation step was critical for separating the supernatant, which contained the analytes of interest.
The supernatant obtained was carefully extracted and stored at − -20 °C for future analyses. This freezing step is crucial for preserving the integrity of the substances contained within the supernatant.
To specifically measure the concentrations of certain cytokines, namely TNF-α, IL-1β, and IL-12, enzyme-linked immunosorbent assay (ELISA) kits were used. These kits, sourced from Abcam (Cambridge, UK), were designed for precise cytokine quantification. These kits were used in strict accordance with the manufacturer's guidelines and procedures, ensuring the standardization and reliability of the results. By following these detailed protocols, this study aimed to accurately determine the levels of these cytokines, which are key indicators of inflammatory responses in colon tissue samples.
Quantitative Real-time Polymerase Chain Reaction (qRT-PCR)
In the quantitative real-time polymerase chain reaction (qRT-PCR) phase of the study, total RNA was extracted from mouse colon tissue samples using the TRIzol reagent. To ensure the quality of the extracted RNA, spectrophotometric measurements were taken at 260/280 nm wavelengths. This step is crucial as it assesses both the concentration and purity of the RNA, which are key factors for accurate downstream analysis.
Once the RNA quality was confirmed, it was then converted into complementary DNA (cDNA) using the Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific, USA). This conversion was essential for the subsequent qRT-PCR process, as cDNA served as the template for amplification.
The qRT-PCR was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems, USA), coupled with the SYBR Green Plus reagent kit (Roche, UK). SYBR Green is a widely used dye in PCR assays because it binds to double-stranded DNA and allows for the detection of amplified gene products in real time. The target mRNA molecules for this procedure are TNF-α, IL-1β, IL-12, ZO-1, occludin, and β-actin. β-actin mRNA was included as an internal standard, serving as a baseline for normalization of the data. Specific primer sequences used for each target gene are listed in Table 1.
Table 1 Oligonucleotide primers used for qRT-PCRWestern Blotting Analysis
In the Western blotting analysis phase of the study, the process began with the homogenization of colon tissue samples, which is essential for breaking down cell structures and releasing proteins. Total protein was extracted from homogenized samples. A Pierce BCA protein assay kit (Thermo, USA) was used to determine the concentrations of the extracted proteins. After quantifying the protein content, the protein lysates were separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA, UK).
To prevent nonspecific binding of antibodies to the membrane, blocking was performed using a buffer containing 5% non-fat milk for 2 h. Following blocking, the membranes were incubated overnight at 4 °C with primary antibodies targeting the specific proteins of interest: PPARγ, p-P65, p-IkB, NLRP3, ASC, IL-1β, and caspase-1 (Abcam, UK).
After primary antibody incubation, membranes were incubated with the corresponding secondary antibodies for 1 h at room temperature. Blots were detected using a western blotting detection system (ChemiDoc, Bio-Rad, Hercules, CA, UK) and the intensity of the bands was quantified using image J software. For accurate normalization of the results, β-actin (Abcam, Cambridge, UK) was used as an internal control.
Assessment of Mucosal Barrier of Colonic Epithelial Cells
In this study, quantification of occludin and ZO-1 levels in colonic tissues was performed using qPCR and commercially available ELISA kits (Mybiosource, CA, USA). These kits were specifically designed to accurately measure these proteins. Adhering to the manufacturer's guidelines is crucial to ensure the precision and reliability of the results. Occludin and ZO-1 are important, as they are key markers of intestinal barrier integrity, and their quantification provides valuable insights into the condition of colonic tissues in the context of this study.
Statistical Analysis
The data were statistically analyzed using GraphPad Prism software. To evaluate differences in mean values of normally distributed data, one-way ANOVA was utilized, complemented by Dunnett’s test for post hoc analysis. Statistical significance was determined at a p-value threshold of ≤ 0.05, indicating a significant difference between the compared groups or conditions.



留言 (0)