
記住我
A comprehensive analysis identified a total of 2314 genes exhibiting differential expression, with 1508 genes being upregulated and 806 genes being downregulated. The selection of these differentially expressed genes (DEGs) was based on criteria of |log2FC|> 1 and adj. p < 0.05, as illustrated in (Fig. 1A). Furthermore, Fig. 1B presented a heatmap showcasing the top ten significantly upregulated and downregulated genes.
Fig. 1
DEGs identification and Immune infiltration analyses. A, B The volcano plot and heatmap are used to show the top 10 up- and 10 down-regulated genes among the 2314 DEGs. C The distribution of 22 distinct subpopulations of immune cells is observed in each ccRCC sample. D A comparison is made between the immune cell infiltration scores of ccRCC patients and a control group. E Intrinsic correlation of 22 infiltrating immune cells in ccRCC
3.2 Immune infiltration analysesFigure 1C were employed to ascertain the proportional representation of 22 distinct subgroups of immune-infiltrating cells in each sample. A heatmap was created to summarize the ratings of immune cell penetration in individuals diagnosed with ccRCC and the comparison group (Fig. 1D). To obtain a deeper understanding of the possible inherent connections between invading immune cells, we conducted a correlation analysis to visually represent their extensive interactions (Fig. 1E). According to our analysis, a significant negative correlation (cor = − 0.72) exists between CD4+ memory resting T cells and CD8+ T cells.
3.3 Weighted co-expression network construction and identification of key modulesThe samples were clustered using the Pearson’s correlation coefficient. After removing any outliers, a cluster tree was generated for the remaining 584 samples (Fig. 2A). Figure 2B shows the application of a soft threshold of five in order to build a scale-free network. Using average hierarchical clustering and dynamic tree clipping, we were able to identify a total of nine modules (Fig. 2C). In the Fig. 2D, there was a significant association between the green module and γδ T cells. To ascertain relevant genes, a screening process was undertaken to identify genes with a gene significance (GS) value exceeding 0.2 and module membership (MM) value surpassing 0.5 (Fig. 2E). By overlapping the 699 module genes with the DEGs, we were able to acquire 304 DEGRGs (Fig. 2F). Furthermore, we identified the top 20 mutated DEGRGs in ccRCC with ranked percentages, and found increased somatic mutations in CDCA2, NLRC5, DOCK2, KIF21B, and MKI67 (Fig. 2G).
Fig. 2
Construction of weighted co-expression network and analysis of DEGRGs. A Cluster tree for the 584 samples. B The ideal soft threshold for evaluating a network of scale-free co-expression. C A dynamic tree-cutting algorithm was used to combine related genes with comparable expression patterns into one module, resulting in a hierarchical clustering tree. D Heatmap of the correlations between the modules and immune-infiltrating cells (traits). E Module Membership in green module. F Venn diagram of DEGRGs. G The top 20 mutated genes of DEGRGs in ccRCC. GO H and KEGG I enrichment analysis of DEGRGs
3.4 GO and KEGG enrichment analyses of DEGRGsGO functional analysis of the 304 DEGRGs identified involvement in T cell activation, regulation of T cell activation, and leukocyte cell–cell adhesion (Fig. 2H). KEGG pathway analysis of the 304 DEGRGs indicated significant associations with cytokine–cytokine receptor interactions, chemokine signaling pathways, and viral protein interactions with cytokines and cytokine receptors (Fig. 2I).
3.5 Establishment and validation of the prognostic modelAnalysis of the 304 DECRGs using univariate Cox regression revealed that 70 DEGRGs displayed a significant association with the OS of ccRCC patients (p < 0.2) (Fig. 3A). In order to reduce the effects of overfitting, we employed LASSO analysis to choose the most significant genes for our predictive model. We discovered and kept 13 genes that showed substantial predictive worth (Fig. 3B, C). Afterwards, we created a risk score equation for ccRCC patients by utilizing the expression levels of the previously mentioned 13 genes. The risk score can be calculated using the following formula: risk score = (0.159 × PLCB2 expression value) + (0.141 × TMSB10 expression value) + (0.055 × ZNF80 expression value) + (0.099 × IL4I1 expression value) + (0.058 × TNIP3 expression value) + (0.011 × C2 expression value) + (0.043 × PMCH expression value) + (0.241 × NUSAP1 expression value) + (0.032 × HSD3B7 expression value) + (0.030 × BATF3 expression value) + (− 0.178) × ENPP3 expression value) + (0.033 × NNMT expression value) + (0.005 × LHFPL2 expression value).
Fig. 3
Establish and verify the prognostic model. A Univariate Cox regression analysis. B, C LASSO analysis with a minimum lambda value. In the training set: D, E Risk score and the survival status F Kaplan–Meier curves of the high- and low- risk groups (p < 0.0001). G ROC curve for 1-, 3-, 5-year survival predictions in patients with ccRCC based on the risk score. In the validation set: H, I Risk score and the survival status. J Kaplan–Meier curves of the high- and low-risk groups (p < 0.019). K ROC curve for 1-, 3-, 5-year survival predictions in patients with ccRCC based on the risk score
The risk score for each patient in the training and validation sets was computed by utilizing the established formula. Following this, we categorized them into either the low-risk or high-risk group based on the median risk score. The distribution of the risk score and survival status among the high-risk and low-risk groups is illustrated in Fig. 3D, E within the training set. The visual depiction offers a comprehensive comprehension of the distribution of risk scores and their correlation with the patients' survival results. Based on the findings from the Kaplan–Meier analysis, it was observed that the high-risk group demonstrated a significantly diminished OS in comparison to the low-risk group, as depicted in Fig. 3F. Furthermore, the ROC analysis provided evidence of the risk score's robust predictive ability in determining the survival status. The ROC curve demonstrated the effectiveness of the risk score in accurately predicting the survival status, as evidenced by the AUC values of 0.715, 0.69, and 0.678 for 1-, 3-, and 5-year survival, respectively (Fig. 3G). The distribution of risk scores and survival statuses within the high- and low-risk groups of the validation set is depicted in Fig. 3H, I. The Kaplan–Meier analysis demonstrated a statistically significant disparity in overall survival between the high-risk and low-risk groups (Fig. 3J). The ROC curve showed that the risk score performed well in predicting survival status with AUC values for 1-, 3-, and 5-year survival of 0.821, 0.754, and 0.731, respectively (Fig. 3K).
3.6 Clinical feature analysisSignificant correlations were found when assessing the connections between the risk score and different clinicopathological characteristics, including age, gender, laterality, grade, pathologic T, pathologic M, pathologic N, and tumor stage. Significantly, the risk score showed noteworthy correlations with grade, pathologic T, pathologic M, pathologic N, and tumor stage (Fig. 4A–E). In contrast, there were no notable connections found between the risk score and gender, age, or laterality (Fig. 4F–H).
Fig. 4
Clinical feature analysis and construction of the nomogram in ccRCC. The relevance between the risk score and clinicopathological traits, including grade (A), pathologic T (B), pathologic M (C), pathologic N (D), tumor Stage (E), Gender (F), age (G) and laterality (H). I Univariate analyses to detect independent prognostic factors. J Multivariate analyses to detect independent prognostic factors. K Nomogram was established. L The calibration curves for 1-, 3-, and 5-year. The data were presented as mean ± SD. ****P < 0.0001
3.7 Construction of the nomogram in ccRCCUnivariate analysis was conducted using age, sex, risk score, pathologic T, pathologic M, pathologic N, and tumor stage as variables (Fig. 4I). The prognostic model was constructed through the implementation of a multivariate analysis, wherein variables such as risk score, age, pathologic N, and tumor stage were taken into account (Fig. 4J). Based on these factors, a nomogram was developed to estimate the survival probability of ccRCC patients at 1-, 3-, and 5-years (Fig. 4K). Figure 4L displayed strong agreement between the forecasted survival probability from the nomogram and the real survival results for the 1-, 3-, and 5-year durations, as shown by the calibration curves.
3.8 Immune analysis of the high- and low-risk groupsThe analysis of the high-risk and low-risk groups showed that the high-risk group had a considerably greater immune score compared to the low-risk group (p < 0.001) (Fig. 5A). Nevertheless, there existed a notable disparity between the two factions (Fig. 5B). By utilizing CIBERSORT, we successfully detected variations in the composition of 15 immune cell categories between the high-risk and low-risk cohorts. The immune cells comprised of inexperienced B cells, plasma cells, T cells with CD8+ markers, T cells with CD4+ markers in a state of memory rest, T cells with CD4+ markers in an activated memory state, T cells that assist in follicular development, regulatory T cells (Tregs), T cells with γδ markers, inactive NK cells, monocytes, macrophages in M0 state, macrophages in M1 state, macrophages in M2 state, dendritic cells in an activated state, and mast cells in a resting state (Fig. 5C).
Fig. 5
Immune analysis of the high and low risk groups and risk model predictability in immunotherapy response immune analysis of high and low risk groups. Immune (A) and stromal (B) scores of high and low risk groups. C Screening immune cell between the high and low risk groups by CIBERSORT databases. D The expression of immune checkpoints between high-risk and low-risk groups. E–G Association between risk model and immune infiltration. H IPS between high and low risk groups. The data were presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
In addition, various immune checkpoint markers, such as BTLA, NRP1, LAIR1, TNFSF4, CD244, LAG3, ICOS, CD40LG, CTLA4, CD48, CD28, CD200R1, ADOR3DL1, CD80, LGALS9, TNFSF14, IDO2, TMIGD2, HHLA2, TNFSF18, CD70, TNFSF9, TNFRSF8, CD27, TNFRSF25, CD40, TNFRSF18, TIGIT, CD86, CD44, and TNFRSF9, showed significant differences in expression levels, comparing immune checkpoint expression in high-risk and low-risk groups (Fig. 5D).
3.9 Predictive potential of the risk model for immunotherapy responseTo enhance our understanding of the predictive capacity of our risk model in anticipating the efficacy of immunotherapy, we conducted an analysis to examine the correlation between the risk model and established indicators of immunotherapy response, such as TIDE and IPS. TIDE scores were strongly correlated with risk scores in the analysis (p < 0.05). Furthermore, we observed a stronger positive association between the risk scores and TIDE dysfunction (R = 0.51, p < 0.05). On the other hand, there was a negative correlation between the risk scores and TIDE exclusion (p < 0.05) (Fig. 5E–G). Furthermore, there was no notable disparity in the IPS observed between the high-risk and low-risk categories. The suggestion is that our risk model may not be directly associated with the efficacy of immune checkpoint inhibitors in this particular situation (Fig. 5H). To summarize, the aforementioned findings indicate that individuals with reduced risk scores are more likely to experience advantages from immunotherapy.
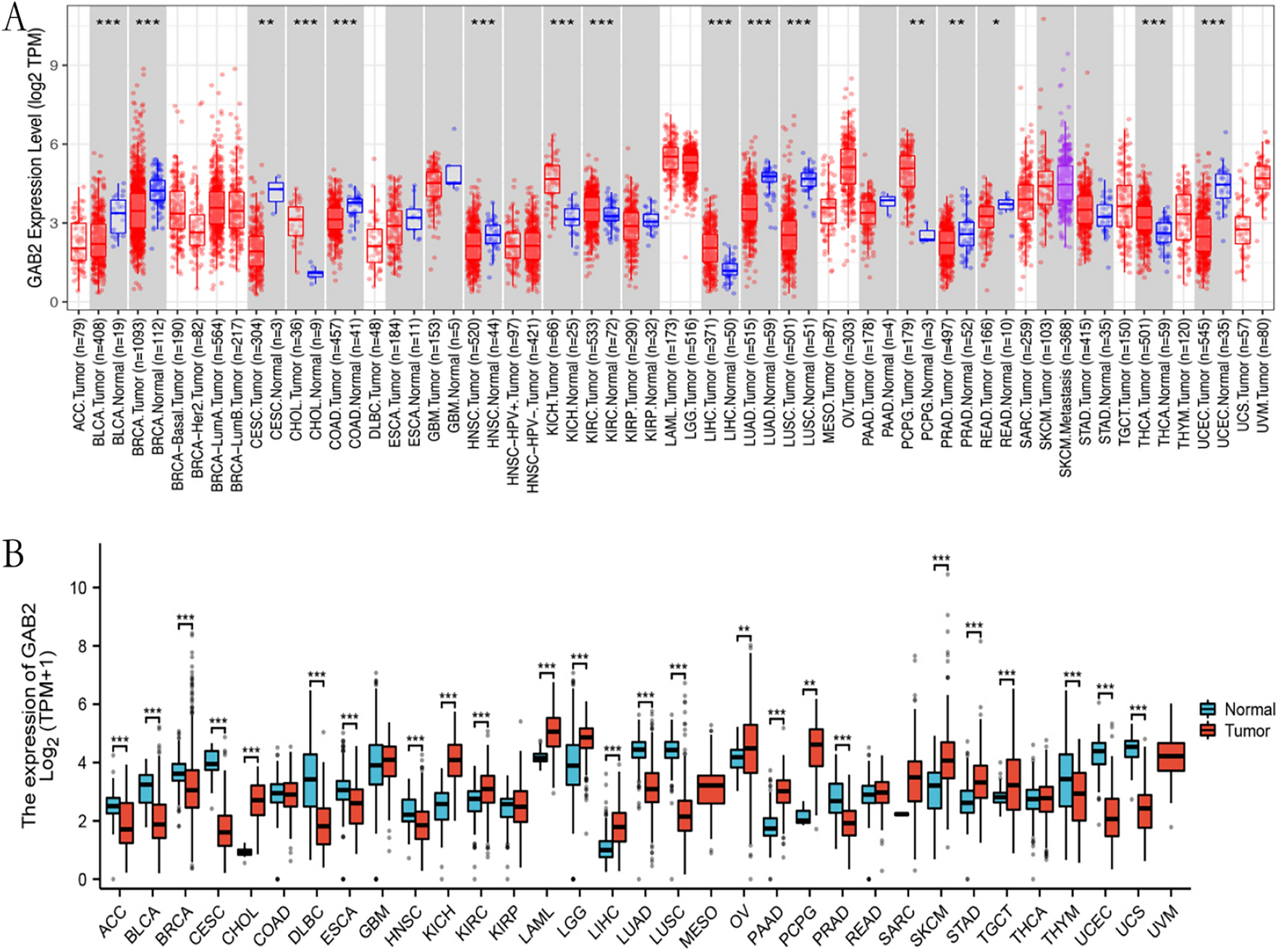
3.10 Potential function of TMSB10 in prognosis prediction, mechanism of immune infiltration, and therapeutic responseGiven the unclear prognostic significance of TMSB10 as a gene related to T cells in ccRCC, we conducted further analyses to investigate the biological function of TMSB10.By analyzing TCGA datasets, we discovered a notable increase in the expression of TMSB10 in tumor tissues compared to normal tissues (Fig. 6A). The study on the survival of ccRCC patients with different levels of TMSB10 expression aimed to assess the predictive ability of TMSB10. The results indicated that higher TMSB10 expression (p < 0.05) was linked to shorter overall survival (OS) based on the analysis (Fig. 6B). To elucidate the biological functions of TMSB10 in immune infiltration, we utilized the TIMER database to examine the association between TMSB10 expression and the abundance of infiltrating immune cells. It was discovered that arm-level deletions accounted for the majority of mutations in B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophils, and dendritic cells (p < 0.001) (Fig. 6C). Moreover, there was a strong correlation between macrophage presence and expression of TMSB10 (p < 0.01) (Fig. 6D). Furthermore, we conducted an assessment of the impact of TMSB10 knockdown on the secretion of immunosuppressive factors TGF-β1 and IL-35 in ccRCC cells using ELISA. The findings indicated that the downregulation of TMSB10 led to a decrease in the secretion of immunosuppressive factors TGF-β1 and IL-35 in ccRCC cells, as illustrated in Figure S1. Consequently, it is suggested that TMSB10 may have a significant involvement in the immune evasion and tumor advancement of ccRCC.
Fig. 6
The clinical significance of TMSB10 in ccRCC. The upregulation of TMSB10 in ccRCC samples, as evidenced by data from TCGA (A) and cell lines (B), is associated with a significantly lower expression level of TMSB10, which in turn is correlated with a favorable prognosis. C Additionally, the copy number of immune cells in ccRCC. D The correlation analysis of TMSB10 with infiltrating B cells, CD4+ T cells, CD8+ T cells, Macrophages, Neutrophils, and Dendritic cells using TIMER further support these findings. The data were presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
3.11 Effect of TMSB10 downregulation on proliferation, migration, and invasion of ccRCC cells in vitroWe suppressed the expression of TMSB10 mRNA in three ccRCC cell lines to understand the biological role of TMSB10. In qRT-PCR assays and Western blots, siRNA transfection remarkably reduced TMSB10 mRNA and protein expression (Fig. 7A, B). TMSB10 knockdown significantly decreased the proliferation of ccRCC cells in CCK-8 assays (Fig. 7C). The findings from the EdU assay also indicated that the suppression of TMSB10 greatly impeded the growth of ccRCC cells (Fig. 7D). Moreover, the Transwell assay findings unequivocally exhibited a significant reduction in the migratory and invasive capabilities of ccRCC cells after TMSB10 knockdown (Fig. 7E, F). These results demonstrate that the proliferative, migratory, and invasive properties of ccRCC cells are inhibited by TMSB10 knockdown.
Fig. 7
The clinical significance of TMSB10 in ccRCC and in vitro study. A, B TMSB10 or a negative control siRNA were transfected to verify transfection efficiency. C CCK-8 proliferation analysis of the effects of TMSB10 knockdown on the cell growth of 786-O and 769-P at 0, 24, 48, 72 and 96 h post-transfection. D EdU assays. E, F TMSB10 knockdown cells were applied for transwell analysis. Representative images of migrated cells and invaded cells were shown. The indicated migrated and invaded cells were quantified in three randomly chosen fields and presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
留言 (0)