
記住我










Glomus tumors (paragangliomas) are rare, benign neuroendocrine tumors that arise in the paraganglionic tissues of the autonomic nervous system.[1] Head-and-neck paragangliomas (HNPGLs) are divided according to which skull base structure or cranial nerve (CN) they are associated with. Glomus jugulare is found near the skull base in the region of the jugular bulb. It arises from the Jacobson nerve (branch of CN IX) or Arnold nerve (branch of CN X) within the jugular foramen.[2] Jugulotympanic paragangliomas include jugulare and tympanicum glomus tumors. Glomus tympanicum occurs in the region of the tympanum and arises from the Jacobson nerve in the middle ear or cochlear promontory.
Glomus tympanicum cases are generally treated surgically. However, glomus jugulare tumors (GJTs) are located in the lateral skull base and, hence, pose a significant surgical challenge.[2] Given the risk of cranial neuropathy with surgery, radiation is preferable. Therefore, management options are observation, surgery, radiotherapy, or a combination of the latter two. One concern is the development of new or worsening cranial neurologic deficits. Surgery is recommended for young patients, secretory lesions, progression after radiotherapy, or malignant transformation.[2]
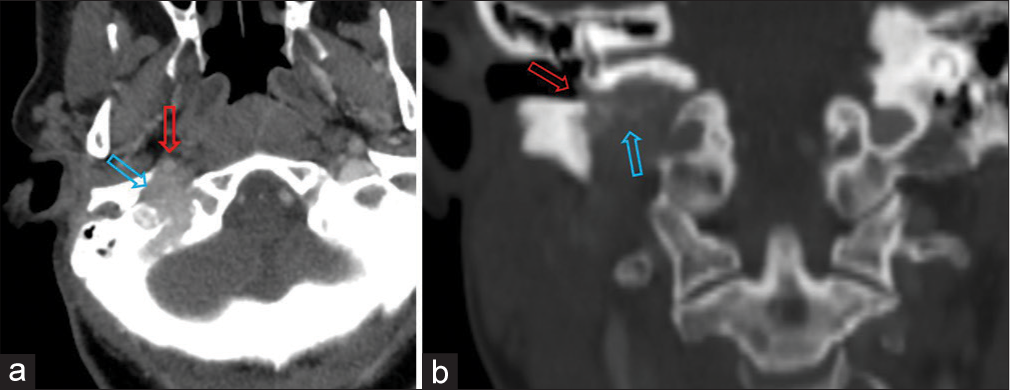
CASE REPORTA 62-year-old female with multiple comorbidities presented to an outside otolaryngologist complaining of otalgia. In December 2022, computed tomography (CT) imaging of the neck showed an ill-defined enhancing lesion of the right jugular fossa with associated bony erosive changes of the inferior petrous temporal bone and jugular spine [Figure 1]. Further, evaluation with magnetic resonance imaging showed a lobulated T2 iso to mildly hyperintense mass lesion with intralesional flow voids centered in the right jugular foramen with herniated and erosive changes of the surrounding bone and occlusion of the right jugular bulb, consistent with a paraganglioma [Figure 2]. Notably, it showed superolateral extension of the paraganglioma to involve the right hypotympanum/middle ear cavity and also mild anterior extension into the vertical petrous carotid canal secondary to the destruction of the right carotid, jugular spine. Superiorly, it involved the contiguous right petrous temporal bone and posteriorly involved the adjacent right mastoid bone and right occipital squamosal bone, and medially extended into the right hypoglossal canal and inferiorly extended into the superior right carotid space. Based on these findings, diagnosis of a glomus jugulare tympanicum was made, requiring consideration of a potential surgical approach for resection. A comprehensive head and neck examination showed a red hue at the inferior aspect of the right tympanic membrane. The flexible fiberoptic laryngoscopy was completely normal.
Export to PPT

Export to PPT
The decision was made to pursue evaluation by neurotology, endocrinology, and radiation oncology. During the neurotology evaluation, the patient reported several months of increasing difficulty hearing in the right ear (“plugged”). She denied vertigo, dysphonia, dysphagia, or any facial weakness. The Weber test lateralized to the right; however, the Rinne test was normal. Otoscopic examination showed a vascular lesion along the posterior inferior aspect of the right middle ear. The patient was diagnosed with a right GJT which clearly extended into the middle ear. Considering that the patient did not have any CN deficit, and the risk of cranial neuropathy with surgical intervention, radiotherapy was recommended. The patient was offered testing for succinate dehydrogenase genetic variations to rule out hereditary paraganglioma, which she declined. She also declined audiogram testing. Then, the patient was seen by endocrinology, and appropriate laboratory work was ordered to further characterize her case. Her chromogranin A and plasma metanephrine were both normal. However, plasma normetanephrine was very slightly elevated at 0.96 nmol/L (range 0.0–0.89 nmol/L). Thyroid-stimulating hormone and free T4 were both within the normal range.
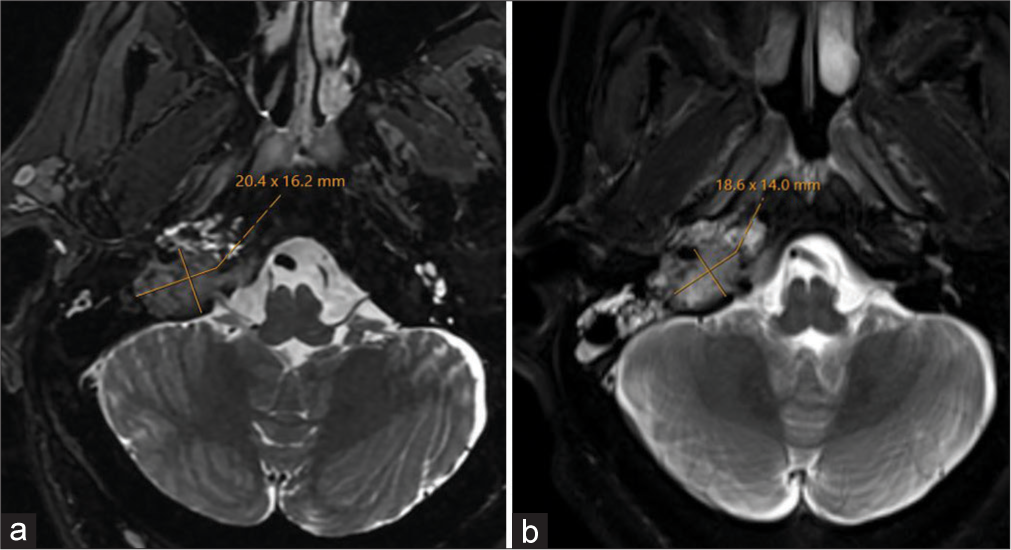
After discussion with our neurologic specialist radiation oncology colleagues, the decision was made to proceed with a course of conventional fractionation (50.4 Gray in 28 fractions) volumetric modulated arc therapy. The patient started the course of radiotherapy in late April 2023 and went through treatment with very few acute toxicities or side effects. There were no breaks in the treatment course or any changes to the treatment plan. Follow-up imaging showed T2 isointense to mildly hyperintense mass lesion with intralesional flow voids centered in the right jugular foramen with permeative and erosive changes of the surrounding bone and occlusion of the right jugular bulb, with overall size slightly decreased [Figure 3]. Recently, the patient was diagnosed with non-small cell carcinoma of the lung, and at the time of this manuscript submission, is undergoing treatment.
Export to PPT
DISCUSSIONHNPGLs account for around 3% and also represent <0.5%, of all head-and-neck cancers.[3] Most HNPGL cases arise from the jugular bulb, carotid body, or some lower CNs such as the glossopharyngeal or vagus nerves, and are named accordingly.[2] Head-and-neck glomus tumors are rare neuroendocrine tumors which can be addressed by observation, surgery, radiotherapy, or a combination of the latter two. The Fisch classification of jugulotympanic tumors is based on the extent of local spread and involvement of surrounding structures and should be considered. Type A refers to cases that arise along the tympanic plexus and are limited to the middle ear cleft. Type B is cases that invade the hypotympanum but has no infralabyrinthine component (no bony erosion). Type C includes cases that invade and cause bony destruction of the infralabyrinthine component of the temporal bone. Type D cases have varying degrees of intracranial extension. Our patient was considered a Type C case due to extension into the vertical carotid canal.
Approximately 30% of these head-and-neck tumors occur in the jugulotympanic region, 57% in the carotid body, and 13% in the vagus nerve.[4] Chronic hypoxia is the only known acquired risk factor.[5] While most cases are benign, an estimated 6–19% of HNPGLs develop metastases in regional lymph nodes, though distant metastases can occur.[6] Most cases are sporadic, though paragangliomas can develop due to germline sequence variations in one of more than 15 different susceptibility genes, somatic sequence variations, or fusion genes.[7]
As predicted by the indolent course of disease, symptoms often develop gradually and are dictated by the location of the primary tumor. Middle ear involvement causes conductive hearing loss, tinnitus, and a feeling of ear fullness; intracranial extension causes nausea and headache; and CN involvement leads to speech and/or swallowing dysfunction.[2] Glomus vagale cases often present as a neck mass or with hoarseness. Jugular foramen tumors can present with hearing loss, pulsatile tinnitus, dysphonia, or even shoulder weakness.
Management decisions depend on a number of factors, including age of the patient, risk of malignancy, and location of the tumor (size, shape, and rate of growth). Options are observation (with close follow-up), surgery, and radiotherapy. While surgery or combination offers great chance of controlling the disease, given the risk of cranial neuropathy with surgery, both can cause significant short- and long-term morbidity. Hence, there has been a trend in recent years toward managing patients with observation, or using radiation as the preferred treatment for growing tumors with control criteria of the disease through radiotherapy being (1) absence of progression of symptoms or CN dysfunction, and (2) the lesion did not increase in size according to physical examination or radiological control.[8] One group studied 47 cervical paragangliomas in 43 patients and found that at a follow-up of 5 years, 42% of cases had remained stable, 38% grew, and 20% regressed.[9]
A recent systematic review and meta-analysis looked at 852 jugulotympanic tumors across nineteen studies and compared radiosurgery to surgical resection.[10] There was a non-significant tumor recurrence rate of 3.5% after radiosurgery compared to 3.9% after surgery. However, the complication rate was worse after surgery (29.6%) versus radiosurgery (7.6%). While focused on radiosurgery, this study does demonstrate efficacy and relative safety of radiotherapy in jugulotympanic cases.
CONCLUSIONGlomus tumors of the head and neck are rare neuroendocrine tumors which pose a difficult decision due to location and potential for malignant transformation. Modern radiotherapy is an effective and often safe non-surgical treatment modality for such patients.
留言 (0)