Primary DIPG and pcGBM culture
Patient-derived primary DIPG and pcGBM cultures (SU-DIPG-XIII-FL, SU-DIPG-XIII-P, SU-DIPG-VI, SU-DIPG-XIX, and SU-pcGBM2), obtained at the time of biopsy or autopsy, were provided by Dr. Michelle Monje at Stanford University. All human tumor cell cultures were generated with informed consent and under institutional review board (IRB)-approved protocols, as previously reported [3, 4, 8]. All patient-derived cultures were grown as tumor neurospheres in tumor stem medium consisting of DMEM/F12 (Invitrogen, 11330032), Neurobasal(-A) (Thermo Fisher Scientific, 10888022), B-27 supplement without vitamin A (1:50, Thermo Fisher Scientific 12587010), human EGF (20 ng ml−1, Shenandoah Biotech 100–26), human b-FGF (20 ng ml−1, Shenandoah Biotech 100–146), human PDGF-AA (10 ng ml−1, Shenandoah Biotech 100–16), human PDGF-BB (10 ng ml−1, Shenandoah Biotech 100–18), and heparin (2 µg ml−1, StemCell Tech 07980). Media was changed once per week. See Additional file 1: Table S1 for details of the patient-derived cultures used in this study.
hiPSC culture
All human induced pluripotent stem cells (hiPSCs) were previously validated with respect to their stemness and differentiation capacity [28]. All hiPSCs were tested for and maintained mycoplasma free. Approval for this study was obtained from the Stanford IRB, and informed consent was obtained from all donors. hiPSCs were maintained in their pluripotent state by being cultured with mTeSR Plus media (StemCell Tech 100-0276) in monolayer on hESC-qualified Matrigel (0.1 mg ml−1, Sigma 354277).
Neural organoid differentiation and maturation
Dorsal forebrain organoids were differentiated according to previously published protocols [11, 29]. Briefly, hiPSCs were dissociated with Accutase (StemCell Tech 07920) and aggregated into uniform 5000 cell aggregates with AggreWell800 plates (StemCell Tech 34815) in mTeSR Plus media supplemented with ROCK inhibitor Y-27632 (10 µM, Selleckchem S1049). After 24 h, hiPSC aggregates were transferred to ultra-low attachment culture dishes (Corning 4615) in Essential 6 medium (Thermo Fisher A1516401) supplemented with the two dual SMAD inhibitors SB-431542 (10 µM, Tocris 1614) and LDN-193189 (100 nM, StemCell Tech 72147). Media was changed daily. On day 6 of differentiation, neural organoids were transferred to neural medium consisting of Neurobasal(-A) (Thermo Fisher 10,888,022), B-27 Supplement without vitamin A (1:50, Thermo Fisher 12587010), GlutaMAX Supplement (1:100, Thermo Fisher 35050079), Penicillin–Streptomycin (1:100, Thermo Fisher 15070063), and supplemented with human EGF (20 ng ml–1, PeproTech AF-100–15) and human FGF-2 (20 ng ml–1, PeproTech AF-100-18B). From day 25 to 42, neural medium was supplemented with the growth factors BDNF (20 ng ml–1, PeproTech AF-450–02) and NT3 (20 ng ml–1, PeproTech AF-450-03), and media was changed every other day. From day 43 onward, neural organoids were maintained in neural medium with media changes every four days.
Generation of DIPG-neural assembloids
To generate DIPG-neural assembloids, dorsal forebrain organoids (between days 43–80) and DIPG spheroids were generated separately and then assembled by placing them in close proximity at the bottom of a 1.5 mL Eppendorf tube in neural medium in an incubator. DIPG-neural assembloids fused within 24 h and were transferred to 24-well plates using a cut p1000 pipette tip. Media was changed every 4 days.
Assessing DIPG adhesion and migration in 2D
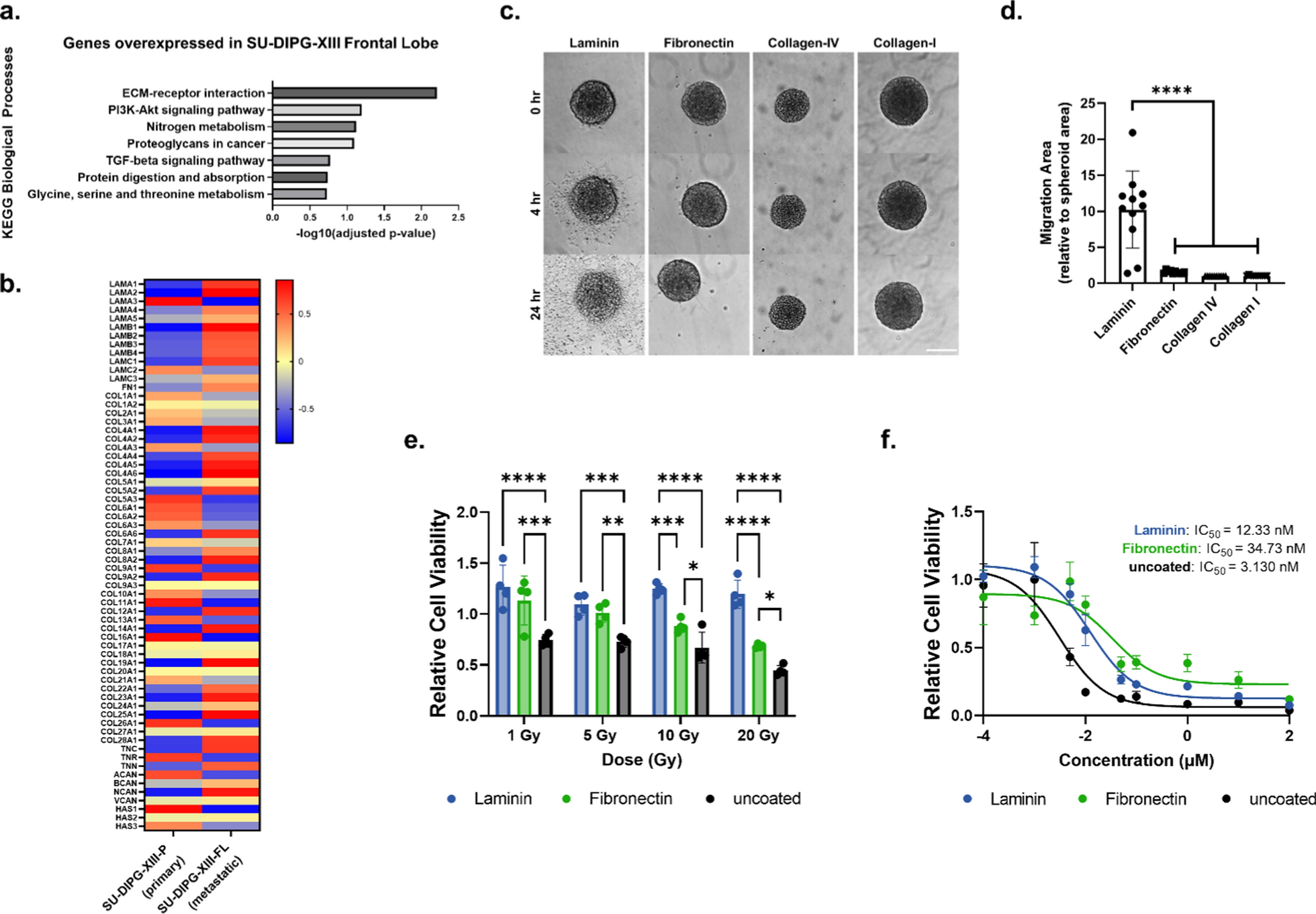
Tissue culture plastic (TCP) was coated with laminin (50 µg mL−1, Sigma-Aldrich L2020), fibronectin (50 µg mL−1, Fisher Scientific CB-40008A), collagen-I (50 µg mL−1, Corning 354236), or collagen-IV (50 µg mL−1, Sigma-Aldrich C0543) solutions for 2 h at 37 °C. Solutions for laminin and fibronectin were dissolved in DPBS; solutions for collagen-I and collagen-IV were dissolved in 0.1% acetic acid. Wells were subsequently washed 3× with DPBS. DIPG spheroids were then placed on coated TCP to assess adhesion and migration. Images were collected with a Keyence BZ-800 with a 10X objective shortly after seeding the DIPG spheroids, 0 h, and at 24 h. To quantify migration, ImageJ was used to measure the migration area at 24 h, which was normalized to the spheroid area at 0 h. For live-cell time-lapse imaging to monitor DIPG adhesion and migration, a DMi8 inverted epifluorescence microscope (Leica) with a 20X objective and equipped with a live-cell incubator was used. Images were captured every 10 or 20 min for 24 h.
Generation of integrin knockdown cell lines and characterization using flow cytometry
shRNA constructs against ITGα, ITGβ1, and ITGαV (Sigma Aldrich) and nontarget scrambled shRNA construct (generously gifted by the laboratory of Dr. Michelle Monje) were packaged into lentiviral particles by the Neuroscience Viral Vector Core at Stanford University. For lentivirus infection, dissociated SU-DIPG-XIII-FL cells were exposed to shRNA-expressing lentivirus for 12–16 h before replacing with fresh medium to allow cells to recover. After 48 h, puromycin (Sigma Aldrich P8833) was added to select positively infected DIPG cells. After puromycin selection, DIPG cells were grown for at least one passage before using in experiments. The shRNA constructs are provided in Additional file 1: Table S2.
To assess knockdown efficacy, SU-DIPG-XIII-FL cells transfected with shRNA-expressing lentivirus were characterized by flow cytometry. Cells were dissociated, permeabilized, and stained for intracellular and extracellular protein expression. The following antibodies were used to assess ITGα6, ITGβ1, and ITGαV expression: ITGα6 (PE-CF594 Rat Anti-Human, BD bioscience 562493), ITGαV (PE anti-human CD51, BioLegend 327909), and ITGβ1 (BV786 Mouse Anti-Human CD29, BD bioscience 564815) expression. Results were analyzed by FlowJo Software.
Radiation and panobinostat treatment on 2D and within DIPG-neural assembloids
For 2D studies, DIPG and pcGBM neurospheres were dissociated, and single cells were seeded on ECM coated 96 well plates at 1000 cells/well and allowed to adhere overnight. Plates were coated with either laminin (50 µg mL−1, Sigma-Aldrich L2020), fibronectin (50 µg mL−1, Fisher Scientific CB-40008A), or laminin and fibronectin (each at 50 µg mL−1). Cell treatment started one day after seeding. For radiation studies, cells were irradiated using a SmART cabinet X-ray irradiator (Precision X-Ray). A single beam was targeted at the cells, delivering the following doses: 1, 5, 10, and 20 Gy. Cells were cultured for an additional 5 days, at which point Presto Blue assay (Thermo Fisher Scientific P50201) was used to measure the cell viability. For panobinostat (Selleckchem S1030) treatment, cells were treated at a concentration range from 0.1 nM to 100 µM. Cell viability was measured at day 3 after panobinostat treatment. Relative cell viability was calculated by normalizing treatment groups to the untreated control.
For radiation treatment of the DIPG-neural assembloids, the assembloids was irradiated at 5 Gy and 20 Gy as described above on day 7. Assembloids were cultured for 6 more days, after which samples were fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences 15700) in DPBS for immunohistochemistry. For panobinostat treatment, DIPG-neural assembloids were cultured first for 7 days, then treated with 200 nM panobinostat for 3 more days before harvest. Samples were fixed in 4% PFA in DPBS for immunohistochemistry.
Immunohistochemistry
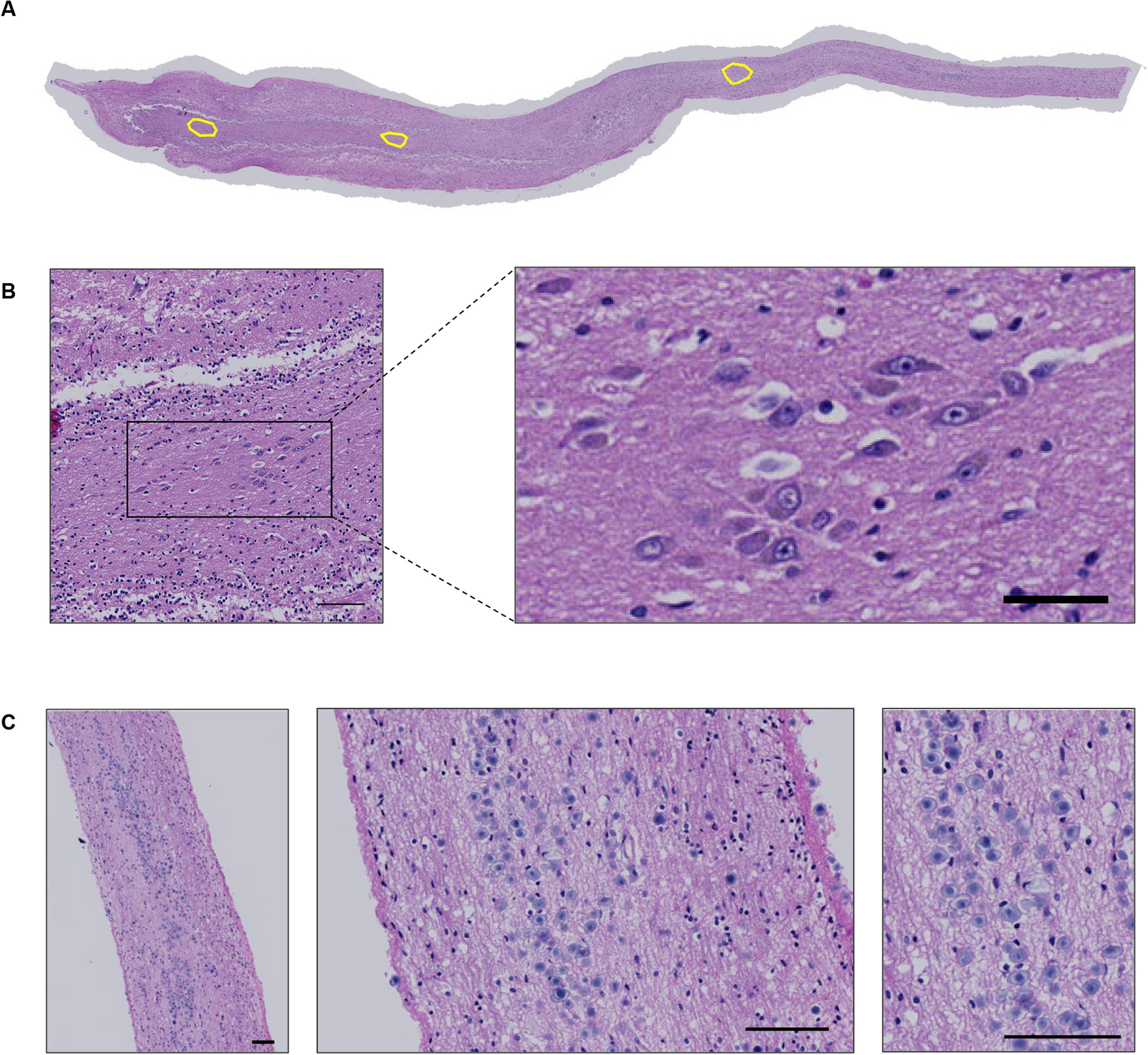
Organoids and assembloids were fixed in 4% PFA for 2 h at 4 °C. They were then washed in DPBS three times for 15 min each and transferred to a 30% sucrose solution in DPBS for 2–3 days at 4 °C. Once the organoids or assembloids sank in the sucrose solution, they were embedded in a 1:1 mixture of OCT (Fisher Scientific 23-730-571) and 30% sucrose in DPBS and snap-frozen using dry ice. For immunostaining, 40 µm sections were cut using a Leica cryostat. Cryosections were washed with DPBS to remove excess OCT and permeabilized with 0.25% Triton X-100 (Thermo Fisher A16046) in DPBS (DPBS-T) for 1 h at room temperature (RT). They were then blocked in 5% goat serum (Gibco 16210-072), 5% bovine serum albumin (BSA, Sigma A9418), and 0.5% Triton X-100 in DPBS for 3 h at RT. The sections were then incubated overnight at 4 °C with primary antibodies diluted in 2.5% goat serum, 2.5% BSA, and 0.5% Triton X-100 in DPBS. To facilitate visualizing infiltrating DIPG cells into the assembloid, DIPG cells were labelled with GFP and also stained with H3K27M, a marker for DIPG. The following primary antibodies were used including GFP (rabbit, 1:200, Thermo Fisher A11122), cleaved caspase-3 (rabbit, 1:400, Cell Signaling 9661), gamma H2A.X (mouse, 1:200, Abcam ab26350), laminin (rabbit, 1:300, Abcam ab11575), fibronectin (rabbit, 1:100, Invitrogen PA1-23693), ZO-1 (mouse, 1:150, Invitrogen 33-9100), histone H3 (mutated K27M) (rabbit, 1:400, Abcam ab190631), Ki67 (rabbit, 1:200, Abcam ab16667). DPBS-T was used to wash the samples three times for 30 min each, and the samples were then incubated overnight at 4 °C with secondary antibodies Alexa Fluor 488 (1:500, Thermo Fisher A-11034) and Alexa Fluor 647 (1:500, Thermo Fisher A-21236) and 4′,6-diamidino-2-phenylindole (DAPI, 5 mg mL−1 stock, 1:2000, Thermo Fisher 62247) in the same antibody dilution solution. The next day, samples were washed with DPBS-T three times for 20 min each and mounted to No. 1 glass coverslips with ProLong Gold Antifade Reagent (Cell Signaling 9071). Samples were imaged using a Leica STELLARIS 5 confocal microscope with a 10X or 63X objective.
RNA-sequencing analysis
Transcriptome data (RNA-seq) of DIPG cell cultures derived from SU-DIPG-XIII-P and SU-DIPG-XIII-FL were downloaded from the GEO dataset with accession ID: GSE99812. SU-DIPG-XIII-P and SU-DIPG-XIII-FL cell cultures used to generate the RNA sequencing data sets were cultured under similar conditions as reported above. Trimmed reads were aligned to the Human reference genome hg38 using the RNA STAR aligner. featureCounts was used to determine read abundance and generate count files. Differentially expressed genes (DEGs) were determined using DESeq2 [27]. DEGs with an adjusted p-value of less than 0.1 were considered significantly different and used for subsequent pathway analysis using the EnrichR tool [28]. Heatmaps depicting gene expression profiles were made using the average z-scores of vst-normalized RNA-seq counts obtained from DESeq2. Count files for the SU-DIPG-VI, SU-DIPG-XXI, SU-DIPG-XXV and SU-pcGBM2 cell lines were downloaded from GEO datasets with accession IDs GSE222481, GSE222560, and GSE99045. TPM (transcripts per million) were extracted and log values were used to generate plots comparing transcript abundance across all cell lines. See Additional file 2: Table S4 for mean gene expression values, log2(fold-change), p-values, and adjusted p-values (False Discovery Rate).
Quantitative polymerase chain reaction (qPCR)
DIPG and pcGBM spheroids were dissociated, immediately resuspended in TRIzol reagent (Invitrogen 15596018), and frozen at −80 °C until use. mRNA was purified from lysates using a phenol–chloroform extraction. Samples were first disrupted by probe sonication (50% amplitude (25 watts), 30 kHz frequency, 0.5 s cycle), transferred to phase lock gels (Quantabio 5PRIME 2302830), and 100 µL of chloroform (Sigma CX1055) was added to each sample. Samples were then centrifuged at 15,300×g for 15 min at 4 °C, and the aqueous phase was transferred to a clean 1.5-mL microcentrifuge tube. Samples were precipitated with isopropyl alcohol and washed twice with 70% ethanol, with centrifugation steps between each wash (18,500×g for 10 min at 4 °C). Samples were then dried and resuspended in 15 µL of nuclease-free water. mRNA concentrations were measured on a NanoDrop (Thermo Scientific), and 800 ng mRNA of each sample was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems 4368814). For each gene target, qPCR was performed on 6.6 µL of diluted cDNA mixed with 0.9 µL of 5 µM forward and reverse primer pair solution and 7.5 µL of Fast SYBR Green Master Mix (Applied Biosystems 4385612). Samples were run on a StepOnePlus Real Time PCR System (Applied Biosystems). Cycle threshold (CT) values were calculated using the StepOnePlus software (v.2.3) and normalized to GAPDH as a housekeeping gene (∆CT). Statistical analysis was performed before transforming to a natural scale, and relative mRNA expression is reported as a geometric mean with standard deviation. See Additional file 1: Table S3 for information about qPCR primers. Melt curves were performed for all primer pairs.
Image analysis
Quantification of CC3 and γH2AX expression: Images obtained from confocal microscopy were processed using a custom Python script to extract the pixel-by-pixel total sum of the red and blue channels that corresponded to the DAPI and CC3 or γH2AX signals, respectively. The reported CC3 or γH2AX expression was calculated by dividing the total CC3 or γH2AX signal by the total DAPI signal and normalizing it to the untreated group in each shRNA integrin knockdown cell line.
Quantification of DIPG infiltration: A custom Python script was used to assess the extent of DIPG infiltration from images obtained from confocal microscopy. Briefly, after performing a maximum intensity z-projection to produce a 2D image, a graphical interface allowed the user to draw a closed boundary around the neural organoid, separating the organoid from the DIPG spheroid. The boundaries were saved for repeated use, and the drawer ensured the use of consistent criteria for delineating the organoid edge. Afterwards, a point-in-polygon test was leveraged to determine which points lie inside the boundary and the minimum Euclidian distance to the boundary for each interior point. Hence, the algorithm assigned each point on the interior a minimum distance from the boundary and subsequently extracted its corresponding intensity for each signal channel. These distances were used to bin the points into equally-spaced concentric shells that signify escalating “levels” of infiltration towards the organoid center. Additionally, a metric for total infiltration either past a certain micrometer distance from the boundary or a percentage distance towards the organoid center were computed. After determining the points that fall within each level or past a certain threshold, the mean intensity of each marker was computed from all points in that group, and the reported infiltration metrics were calculated by normalizing the average eGFP signal to the average DAPI signal. To quantify infiltration by H3K27M staining, a maximum intensity z-projection was performed to produce a 2D image, after which a straight line was drawn orthogonal from the neural organoid boundary to the H3K27M + cell to determine the infiltration distance.
Statistical analysis
Statistical analyses were performed using GraphPad Prism Software. Specifics on statistical tests used and the corresponding p and n values are provided with each of the figure legends.



留言 (0)