Patients
Patient samples were obtained from n = 4 patients diagnosed with localized prostate adenocarcinomas undergoing prostatectomy in the Department of Urology, University Hospital Münster. The local ethical committee approved the study (2007-467-f-S), and all patients gave informed consent. Native prostatic tissue was examined and processed by a pathologist. The prostatic capsules were intact and seminal vesicles, apex and vesical parts of the prostates were identified. According to preoperative results of prostatic biopsies, samples of prostatic tissue were obtained from anatomical sites of carcinoma-positive biopsy and stored in HBSS buffer (Sigma-Aldrich, Pasching, Germany) at 4 °C until further processing.
Tissue dissociation and initial cell culture
Tissue samples were minced into small (> 2 mm3) pieces and incubated with Dispase/Collagenase IV (Stemcell, Vancouver, Canada) in DMEM/F12 (Sigma-Aldrich) with 5% FCS (Thermo Fisher Scientific, Waltham, MA, USA). On the next day, cells were centrifuged, and pellets were washed with DPBS (Sigma-Aldrich). Afterwards, tissue samples were digested in 5 × Trypsin/DPBS (Sigma-Aldrich) for 1 h at 4 °C followed by 1 U/mL Dispase (Stemcell) in DMEM/F-12 incubation for 2 min. Cells were passed through a 100 μm cell strainer and flow through was resuspended in 3 ml PCPM (DMEM/F12, 5% FCS, 200 mM L-glutamine, Penicillin-Streptamycin, 1 μg/ml Charybdotoxin, 200 μM Hydrocortisone, 20 mg/ml Adenine (all Sigma-Aldrich), 10 μM Y-27632 (Miltenyi Biotec, Bergisch Gladbach, Germany), 12.5 mg/ml insulin (PAN Biotech, Aidenbach, Germany), 1 mg/ml hEGF (Sigma-Aldrich)) and seeded into 12-well plates.
Cloning of immortalization factors into pSBbi vectors
Plasmids (pCMV(CAT)T7-SB100 (#34879), pSBbi-GN (#60517), pSBbi-RP (#60513), pBABE-neo-hTERT (#1774), pBABE-neo largeTcDNA (#1780) were purchased from Addgene. cDNAs were PCR amplified and cloned into the pSBbI vectors via either directional SfiI cloning for hTERT [12] or by cloning using the In-Fusion® HD Cloning Kit (Clontech, Mountain View, CA, USA) for SV40 large T antigen (SV40LT). Primer sequences are listed in Additional file 2: Table S2. Immortalization vectors contained either hTERT or SV40LT under the control of the EF1α promotor as well as a GFP/RFP-2A-puromycin/neomycin selection cassette under the control of the synthetic RPBSA promoter (Additional file 1: Figure S1).
Electroporation of immortalization factors and antibiotic selection
Electroporation followed the Amaxa™ Basic Epithelial Cells Nucleofector™ Kit (Lonza, Cologne, Germany) protocol, using program T0-13. 6 × 105 cells were utilized for electroporation of 2 µg total DNA (0.1 µg pCMV(CAT)T7-SB100 plus either 1.9 µg SV40LT or hTERT plasmids in single experiments or 850 ng of both immortalization plasmids in combinatorial approaches). Upon electroporation, cells were reseeded in 6-well plates. Antibiotic selection was administered 5–7 days post-electroporation using 1 mg/ml G418 (PAN Biotech) and/or 1 µg/ml puromycin (PAN Biotech). G418 was administered for 15–20 days, whereas puromycin treatment lasted for 5–7 days.
Cell culture
The human metastases derived prostate cancer cell lines 22Rv1, LNCaP and PC-3 were purchased from the Leibniz-Institute DSMZ GmbH (Braunschweig, Germany). The human benign prostate epithelial cell line RWPE-1 was purchased from ATCC (Manassas, VA, USA). All cell lines were cultured under matching protocols at 37 °C and 5% CO2. Media was purchased from Sigma-Aldrich. Trypsin–EDTA, phosphate-buffered saline and FCS were purchased from Thermo Fisher Scientific. Hormonal treatment was performed using 10 nM R1881 (Sigma-Aldrich) or DMSO (Applichem, Darmstadt, Germany).
Immunofluorescence and flow cytometry
For immunofluorescence analysis, cells were fixated with 4% formaldehyde (Sigma-Aldrich) in DPBS and permeabilized using 1% Triton-X (Sigma-Aldrich) in PBS. Blocking was performed using 1% BSA (Sigma-Aldrich) in PBS. Primary antibodies were pan-cytokeratin (KRT), clone MNF116, 1:1.00, epithelial cell adhesion molecule (EpCAM), clone EPR20532-225, 1:500 (both Abcam, Cambridge, UK); AR, clone D6F11, 1:600 (Cell Signaling, Danvers, MA, USA) along with respective secondary, conjugated antibodies (1:500, Thermo Fisher Scientific). DAPI (Sigma-Aldrich) was used as DNA staining solution.
For flow cytometry analysis 1 × 105—1 × 106 cells were stained in 0.5% BSA/PBS antibody dilution buffer and labelled with the respective antibodies (CD49f-Pacific Blue, clone GoH3, 1:200; CD26-APC, clone BA5b, both Biolegend, Amsterdam, The Netherlands) and analyzed on a FACS Aria II device. Unstained cells were used as a control. Analysis was performed using FlowJo™ 10.
Analysis of integration and expression of immortalization factors
Genomic DNA (gDNA) was isolated using the NEB Monarch® Genomic DNA Purification Kit (New England Biolabs (NEB), Ipswich, MA, USA). PCR for analysis of integration was performed using primers located within the respective integration cassette. Primers targeting endogenous GAPDH were used as control. For analysis of immortalization factor expression, total RNA was isolated using the RNeasy® Mini Kit (Qiagen, Hilden, Germany) following the manufactures guide. 500 ng of total RNA were reverse transcribed using the Primescript® Reverse Transcription Kit (Takara, Tokyo, Japan). qPCR runs for SV40LT and hTERT expression analysis were performed along with primers for housekeeping genes RPL37A and ACTB. qPCR reactions were run using the PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific) on a QuantStudio 3 cycler (Thermo Fisher Scientific). Primer sequences are listed in Additional file 2: Table S2.
RNA sequencing
Library preparation of total RNA was performed with the NEB Next Ultra II RNA directional Kit and single read sequencing was performed using a NextSeqR® 2000 System with a read length of 72 bp. Using a molecular barcode, the samples were demultiplexed (bcl2fastq2) to fastq data and quality controlled (FastQC). Trimmomatic was used for adapter trimming and read filtering [15]. The resulting reads were aligned to the Ensembl GRCh38 reference genome using Hisat2 [16]. The aligned reads were sorted using samtools [17]. The sorted and aligned reads were counted into genes using htsec-counts [18]. The test for differential expression were performed using the r-package deseq2 [19]. Principal component analysis was executed using ClustVis [20].
Luciferase assay
For determination of AR activity using a luciferase assay, cells were co-transfected with an androgen receptor responsive elements (ARE) Firefly luciferase reporter plasmid (pGL3-4xARE-E4-luc, a gift from Dr. M Carey, Department of Biological Chemistry, UCLA, USA) and Renilla luciferase transfection control plasmid (pRL-TK, Promega, Madison, WI, USA). Subsequently, cells were cultured in presence of either 10 nM R1881 or DMSO.
Dual luciferase reporter assays were performed according to the manufacturer’s protocol using the Dual-Glo® Luciferase Assay System (Promega) on a Varioskan Lux microplate reader (Thermo Fisher). Firefly luciferase activity was normalized to Renilla luciferase activity. Transcriptional activation of Firefly luciferase reporter in R1881 treated cells was presented as luciferase activity relative to the DMSO control.
In vivo experiments
In vivo assays were performed at the Max-Planck Institute for Molecular Biomedicine, Münster, Germany. 5 × 106 cells of MS-pPC-191ST, MS-pPC-192S, MS-pPC-193S, MS-pPC-193ST and 22Rv1 were injected subcutaneously into the nuchal fold of SCID (severe combined immunodeficient) mice along with a 1:1 mixture of PCPM and Matrigel® (Corning, NY, USA). 22Rv1 mice were sacrificed after 2 weeks when the tumor reached a growth of about 2 cm3. MS-pPC-193ST mice were sacrificed after 14 weeks, at this time point three out of four mice displaying a palpable tumor tissue at the site of injection. All other mice were sacrificed after 16 weeks.
Pathological analysis
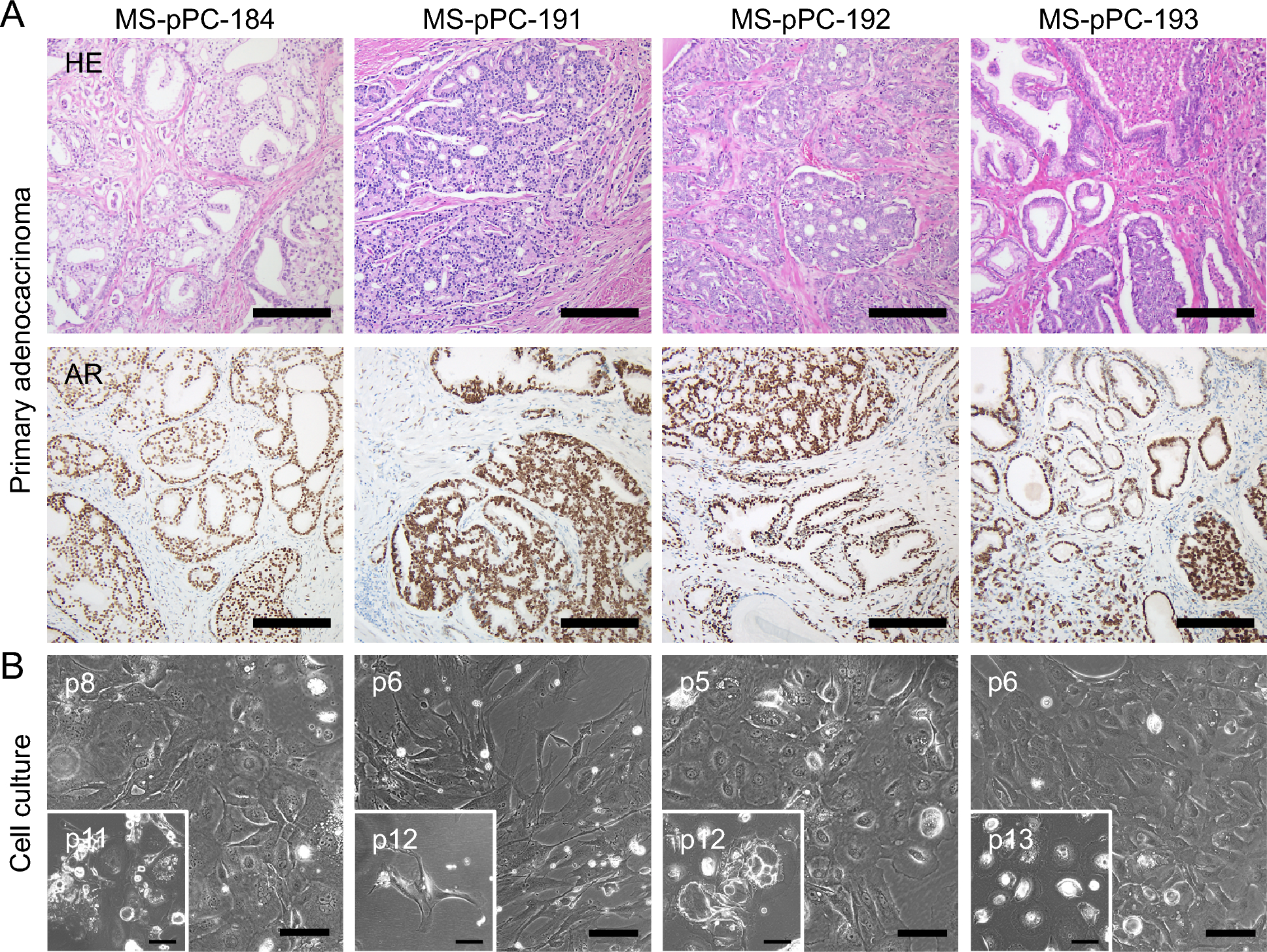
After tumor resection, the specimens were macroscopically examined by a pathologist, fixated in 4% buffered paraformaldehyde and sectioned. Preparation for histological examination, including dehydration, paraffin embedding and hematoxylin and eosin (HE) staining were carried out due to standard protocols at the Gerhard-Domagk-Institute, University Hospital Münster. Histological examination was performed by two pathologists (A.K and S.H.).
Immunohistochemistry was done at the Gerhard-Domagk-Institute using the automated Ventana BenchMark ULTRA IHC staining system (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s instructions. Concisely, 3 µm sections from paraffin-embedded tissue were deparaffinized and pre-treated with Cell Conditioning 1 solution (CC1, Ventana/Roche, Basel, Switzerland) for 24–64 min at 95–100 °C. Subsequently, incubation with primary antibodies (AR, clone SP107, CellMarque, Rocklin, CA, USA; SV40LT, clone PAb101-412, Invitrogen, Waltham, MA, USA) was implemented for 16–32 min at 36 °C. Visualization of immunoreaction was done via Optiview DAB IHC Detection Kit. Tissue sections were counterstained with hematoxylin and blueing solution (all Ventana/Roche).
STR genotyping and authentication
The STR profiling technique took place at the DSMZ under ISO-9001 certified conditions, according to guidelines of the global standard ANSI/ATCC ASN-0002.1-2021 (2021). PCR reaction was performed using 1–2 ng of gDNA with a fluorescent primer set according to ASN-0002.1 and analyzed using capillary separator Genome Lab GenExp Genetic Analysis System (SciEx, Darmstadt, Germany). Allele calling of fragments was carried out by subjecting the reaction products to Genetic Fragment Analyzer software (BeckmannCoulter) and obtained STR profiles were compared for uniqueness in the international STR database using the online tool of the DSMZ (https://celldive.dsmz.de) [21]. A defined search algorithm was also used to check whether cell lines with related profiles or a mix of STR profiles exist. The genetic identity of the established cell lines was authenticated by matching the STR profiles of respective cell lines with generated STR profiles of tumor tissue from FFPE.




留言 (0)