
記住我
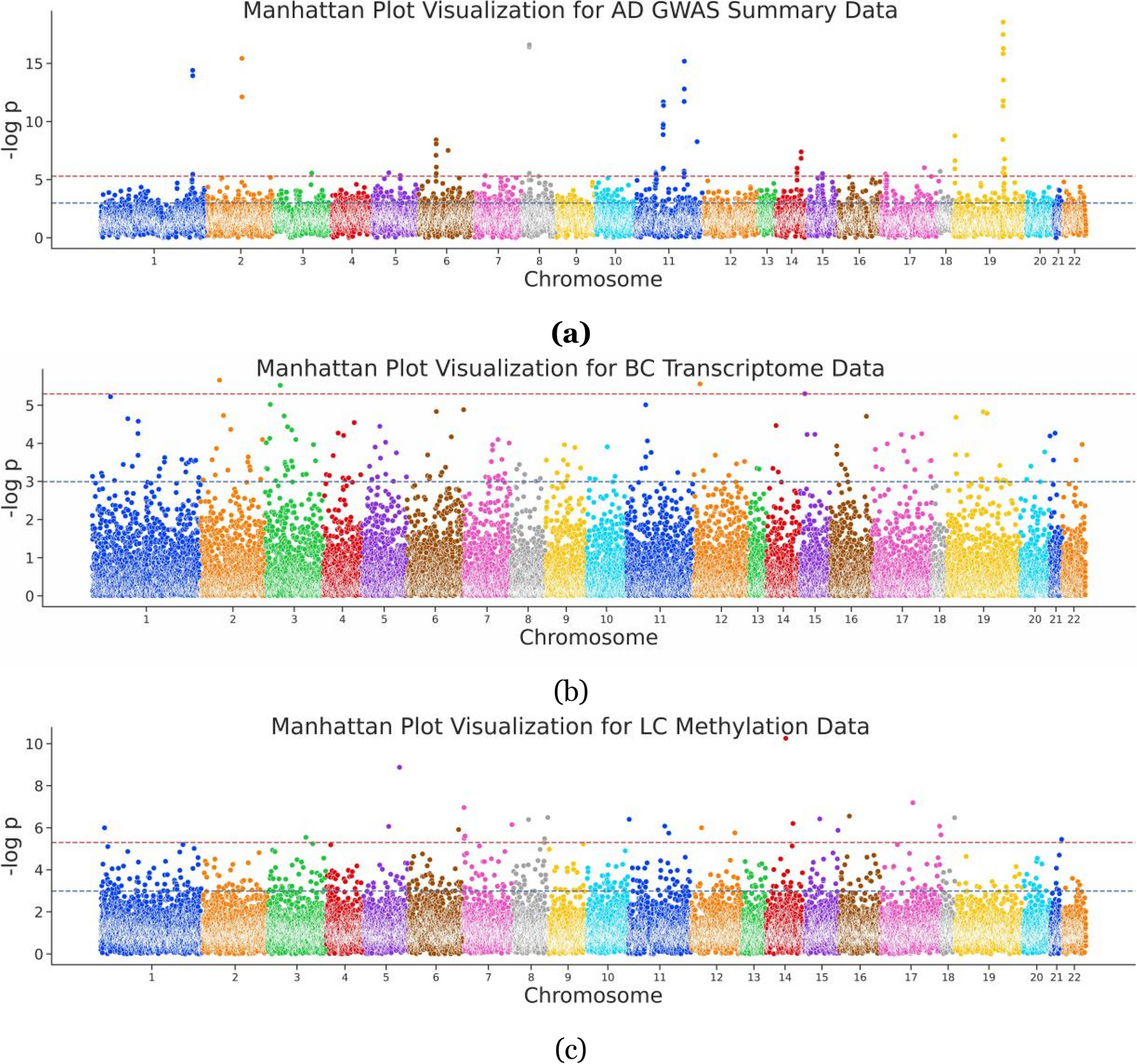
We examined primary, therapy naïve tumor and adjacent non-tumor tissues from AA (n = 31 T, 27 NT) and EA men (n = 32 T and 32 NT) to determine DNA methylation levels in prostate cancer (demographics in Additional File 1, Table S1). Principal component analysis (PCA) showed that DNA methylation, measured as β-value for each CG site (described in the “Methods” section), distinguishes prostate tissues between AA and EA men (Additional File 2, Fig. S2A), based on PC1 and PC2. A PERMANOVA test (10,000 permutations) confirms that tumor samples are significantly different between races (R2= 0.135, pval < 0.0001). PCA did not clearly differentiate between tumor and adjacent non-tumor tissues based on DNA methylation patterns (Additional File 2, Fig. S2A). As a first step to investigate tumor-specific changes in DNA methylation, we established differentially methylated regions (DMRs) using “DMRcate,” an R package [19]. We identified DMRs between tumor and adjacent non-tumor tissues within each race. The top 1000 DMRs, from both race- and tissue- comparisons, are represented in Additional File 2, Fig. S2B, and all DMRs are listed in Additional File 3. An increase in DNA methylation in tumors compared with adjacent non-tumor tissue is referred to as hyperDMRs (top half, represented as −log[10]FDR, in Fig. 1A–B; Additional File 2, Fig. S3A), and a decrease in methylation in tumors compared with adjacent non-tumors is referred to as hypoDMRs (bottom half, represented as log[10]FDR, in Fig. 1A–B; Additional File 2, Fig. S3A). The number of significant hyperDMRs was higher than hypoDMRs in prostate tumors (AA: HypoDMR = 6280/HyperDMR = 11,866 Fig. 1A; EA: HypoDMR = 7715/HyperDMR = 22,042 Fig. 1B). The number of hyperDMRs was twice as low in AA men compared to EA men. These observations, in addition to the PCA plots and lower number of hyperDMRs, suggest that DNA methylation between tumors and adjacent non-tumor tissues is more similar in AA men than EA men. Conversely, we found that adjacent non-tumor tissues from AA men have increased DNA methylation at specific gene regions compared to EA men (red gradient vs blue gradient in adjacent non-tumor tissues, purple rectangles in Additional File 2, Fig. S2B).
Fig. 1
Differentially methylated regions between prostate tumors from AA and EA men reveal race-specific enrichment of pathways and transcript factors. A and B Manhattan plots representing differentially methylated regions in prostate tumors from AA (A) and EA men (B) across all chromosome locations. HyperDMRs are in the top half, represented as −log[10]FDR and hypoDMRs are in the bottom half, represented as log[10]FDR. C and D Enriched pathways associated with DMRs in prostate tumors from AA (C) and EA (D) men. E and F Lisa-derived enrichment of epigenetic regulators enriched in prostate tumors from AA (E) and EA men (F). X-axis: represents hypermethylated regions in prostate tumors, and Y-axis: represents hypomethylated regions in prostate tumors
We assigned DMRs to specific genes based on location and then performed gene set enrichment analysis (GSEA) on these DMR-associated genes to identify specific pathways that were enriched between comparison groups. HyperDMRs were enriched in the PRC2 and H3K27me3 pathways in a race-independent manner (Fig. 1C, D). The association of hyperDMRs with PRC2/H3K27me3-associated pathways was further supported by LISA-based (epigenetic Landscape In Silico deletion Analysis) results. The LISA-based computational algorithm uses ChIP-derived histone marks and chromatin accessibility profiles from Cistrome databases to predict cis-regulatory elements that can regulate gene expression in a given dataset [20]. We applied LISA to the annotated DMRs (AA (n = 31 T, 27 NT) and EA (n = 32 T and 32 NT)) to identify potential transcription factors or co-regulators of gene expression. LISA-derived prediction showed EZH2 enrichment in hypermethylated regions independently of race (Fig. 1E, F). The PRC2 complex primarily methylates histone 3 lysine 27 (H3K27) to repress gene transcription [44]. These observations suggest that the PRC2 complex is present in hypermethylated regions of prostate tumors and can potentially suppress gene transcription. HypoDMRs were enriched in the olfactory and ribosomal pathways in a race-independent manner (Fig. 1C, D). LISA-derived prediction showed enrichment of chromatin-remodeling enzymes, including CTCF and KMT2A, at hypoDMRs of prostate tumors from AA men but not EA men (Fig. 1E, F). HypoDMRs in prostate tumors of EA men were enriched for previously known prostate cancer-associated transcription factors, including FOXA1 and AR [45]. These data support the conclusion that there are interactions between DNA and known prostate cancer-associated transcription factors and chromatin-remodeling enzymes. These interactions may depend on the methylation status of specific genomic regions and are distinct in prostate cancer from AA and EA men.
We also determined race- and tumor-specific changes in DNA methylation using a two-way ANOVA, to account for both race and source tissue (tumor or adjacent non-tumor). This statistical test first determines the methylation difference at each DMR between tumors and adjacent non-tumors within each race, followed by comparisons between AA and EA men (EA(T-NT)-AA(T-NT)). This allowed us to understand the magnitude of DNA methylation change in prostate tumors in relation to adjacent non-tumor tissue, in the context of race. A single DMR annotated to EIF1AY, located on the Y-chromosome, had a greater magnitude of increased methylation in prostate tumors of AA men (P < 0.05, represented as a dot below the second dashed line, Additional File 2, Fig. S3A). This was not surprising as the number of DMRs was much lower in AA men. The positive value resulting from differences in all the other DMRs suggested that prostate tumors from AA men have decreased methylation at specific regions of DNA compared to EA men (gray rectangles Additional File 2, Fig. S2B). These DMRs were annotated to genes crucial for the development of the nervous system, including ZIC1 and EBP41L3 (Additional File 2, Fig. S3A). In agreement with our earlier observations, LISA-derived prediction showed that decreased DNA methylation in prostate cancer was associated with distinct chromatin remodeling enzymes, including BACH1 and PCGF6, in AA men (Additional File 2, Fig. S3B). Our observations suggest that altered DNA methylation, specifically hypomethylated regions, is associated with distinct chromatin-remodeling enzymes in AA men compared to EA men.
Differentially methylated regions in AR target genes and the GATA family of transcription factors are associated with gene expression in a race-specific mannerWe investigated if differential DNA methylation correlates with race-specific changes in gene expression in prostate cancer. We calculated correlation estimates between DMRs and mRNA expression across the entire transcriptome in samples analyzed by both methylation arrays and bulk-RNA sequencing (AA: T = 31, NT = 27, EA: T = 32, N = 31). This allowed us to calculate and compare correlation estimates (ρ) between DNA methylation and gene expression within matched tumor and adjacent non-tumor tissue. We observed positive and negative correlations between DNA methylation and gene expression in tumor versus non-tumor tissues (Additional File 2, Figs. S4A–B, all correlations are listed in Additional File 4). Positive correlation between DNA methylation and gene expression has been established in other models [46]. To our knowledge, our study is the first to report an inverse correlation between Alpha-Methylacyl-CoA Racemase (AMACR) expression and methylation of AMACR-associated DMRs in prostate cancer and adjacent non-tumor tissues (AA: correlation estimate for tumor: − 0.62, P-value: < 0.001; EA: correlation estimate for tumor: − 0.68, P-value: < 0.001; Additional File 2, Figs. S4C–D; Additional File 4). AMACR is a verified biomarker overexpressed in prostate cancer [47]. Compared to adjacent non-tumor tissue, AMACR is overexpressed in prostate tumors from AA men (log2fold change: 1.85, P < 0.001) and EA men (log2fold change: 2.70, P < 0.001) (Additional File 5). Our observations suggest a potential mechanistic link between DNA hypomethylation and AMACR gene expression in prostate cancer.
To identify race-specific associations, we focused on gene expression and DMR correlations that are distinct between AA and EA men. In prostate cancer from AA men, genes including known AR target genes TRIM63, ATP2A1, and ARHGAP28 [48,49,50], showed a significant inverse correlation between gene expression and DNA methylation (Additional File 4). Additionally, we observed race-specific correlations between DNA methylation and gene expression of GATA family members (Fig. 2A–E, Additional File 6). GATA2 and GATA3 are known AR co-regulators in prostate cancer but any race-specific association is unknown [51]. There is also limited knowledge about the role of the other members of the GATA family in prostate cancer [52, 53]. Therefore, we investigated the association between DNA methylation and gene expression of all GATA family members.
Fig. 2
GATA transcription factor DNA methylation and gene expression are associated in a race-specific manner. Each dot represents ρ, correlation coefficient, between methylation and gene expression. Top left quadrant represents a negative correlation between DNA methylation and RNA expression in prostate tumors compared to adjacent non-tumor tissues; i.e., a negative correlation is only found in prostate tumors. Left panels: represent correlation in all prostate tumors. Purple dots represent AA men, and dark cyan dots represent EA men. Middle panels: represent annotated CpG sites in AA men. Right panels: represent annotated CpG in EA men
Regardless of race, GATA3 was significantly downregulated in prostate tumors compared to adjacent non-tumor tissues (Additional File 2, Fig. S4E, P < 0.001). GATA4 was the only member of the family that showed a positive correlation between gene expression and DNA methylation at specific loci in both AA and EA patients and tumor and adjacent non-tumor tissues (Fig. 2C, left panel: right top quadrant, Additional File 6). Conversely, GATA5 and GATA6 gene expression were negatively correlated with DNA methylation at specific loci in prostate tumors and adjacent non-tumor tissues regardless of race (Fig. 2D–E, left panel: left bottom quadrant, Additional File 6).
We identified race-specific differences. There was a significant negative correlation between DNA methylation of the GATA5 transcription start site and RNA expression in tumor tissue of EA men (Fig. 2D, middle and right panels: left top quadrant, Additional File 6). GATA2 expression was negatively correlated with DNA methylation at specific loci in adjacent non-tumor tissues of AA men, suggesting that DNA methylation can regulate GATA2 expression (Fig. 2A, left panel: left bottom quadrant, dots represent correlation coefficient estimate for individual clinical samples, Additional File 6). GATA2 expression inversely correlated with DNA methylation at the multiple CG sites in the 5′-UTR region (P-value: < 0.001) in adjacent non-tumor tissues from AA men (Fig. 2A, middle and right panels: left bottom quadrant, Additional File 6). This suggests that DNA methylation at the 5′-UTR can inhibit transcription and therefore reduce GATA2 gene expression. GATA3 expression negatively correlated with DNA methylation at specific loci in a subset of prostate tumors from AA men (Fig. 2B, left panel: left top quadrant, Additional File 6). Significant correlations of gene expression with DNA methylation were observed explicitly at multiple CG sites at the transcription start site (P < 0.01) in prostate tumors from AA men (Fig. 2B, middle and right panels: left top quadrant, Additional File 6). Additionally, we observed an inverse correlation between DNA methylation and gene expression in annotated GATA5 transcription start sites in adjacent non-tumor tissues from AA men (Fig. 2D, middle and right panels: left bottom quadrant, Additional File 6). Compared to adjacent non-tumor tissues, GATA4 was significantly upregulated in prostate tumors from AA men (Additional File 2, Fig, S3E, P < 0.05). GATA5 expression was significantly downregulated in adjacent non-tumor tissues from AA men compared to EA men (Additional File 2, Fig. S4E). GATA5 was further downregulated in prostate tumors compared to adjacent non-tumor tissues of AA men (Additional File 2, Fig. S4E, P < 0.001). Our findings indicate that epigenetic regulation is an important process, which may impact race-specific expression of genes associated with AR signaling in prostate cancer and adjacent non-tumor tissues.
Patient-specific prostate cancer Boolean network modeling reveals race-specific differences in TGF-β, IDH1, and cell cycle pathwaysTo understand whether differential expression of AR-associated genes (TRIM63, ATP2A1, ARHGAP28, and GATA) indicates dysregulation of AR signaling in general, we first determined AR protein expression in tissue microarrays (TMAs) consisting of prostate tumor tissues and adjacent non-tumor tissues from AA (T: 107, NT: 107) and EA men (T: 133, NT: 133). Given that nuclear AR protein drives transcriptional activity that is important for prostate cancer proliferation and survival [54], we assessed nuclear AR expression. We found race- and tissue-specific correlations between the percent of AR-positive nuclei and clinical parameters, including Gleason score and progression-free survival (Additional File 1, Table S3–4; Additional File 2, Figs. S5A–B). The percent of AR-positive nuclei was higher in adjacent non-tumor but not tumor tissues from AA men compared to EA men (78.20% vs. 73.28%, P < 0.01) (Additional File 1, Table S4; Additional File 2, Figs. S5C–D).
20 samples from AA prostate cancer patients that are present on the TMAs were also analyzed by RNA sequencing. We used these overlapping samples to determine whether AR transcriptional activity is altered. We utilized Gene Set Variation Analysis (GSVA) [42] to derive expression scores for the canonical AR gene targets KLK2, KLK3, NKX3.1, and TMPRSS2 (measured by RNA sequencing) [55, 56]. GSVA scores can be used to determine pathway activity, and in this case AR transcriptional activity, which provides an advantage over single gene measurements. These GSVA scores indicated AR transcriptional activity is significantly higher in prostate tumors than in adjacent non-tumor tissues (P < 0.05) (Additional File 2, Fig. S5E). To add rigor to our study, we expanded our AR activity analysis to the TCGA prostate cancer dataset (EA: n = 270 T, 36 NT; AA: n = 43 T, 6 NT). The results recapitulate findings from the Roswell Park cohort, where we observed an increase in AR transcriptional activity in tumor versus non-tumor in EA but not AA men (Additional File 2, Fig. S5F). To further investigate race-specific differences in AR activity, we performed GSVA for a 27-gene AR activity score developed by Hieronymus, H. et al. [57] in both the Roswell Park and the TCGA cohorts. AR activity was significantly higher in tumor tissues compared to normal tissues from EA men (Additional File 2, Figs. S5G–H). These results show that AR transcriptional activity is significantly higher in tumors from EA men despite AR protein expression being similar in prostate cancer and adjacent non-tumor tissues. Future studies analyzing AR protein expression and AR transcriptional activity from tumor tissues can provide insights into these observations in EA men. These observations suggest that determining AR protein and target gene expression may reflect AR activity more accurately.
Targeting AR to alter transcriptional signaling and block disease progression is a common therapeutic strategy in prostate cancer [58]. It is unknown whether AR targeting in the clinic results in downstream biological consequences that are different for AA and EA prostate cancer patients. To address this question, we adapted an existing prostate cancer Boolean network [15] to analyze a subset of clinical samples from Roswell Park (N = 15, AA: 7, EA: 8; a subset of samples listed in Additional File 1, Table S1). These 15 samples had the three data types (transcriptomics, mutation, and copy number variation) required to generate patient-specific Boolean networks. Men with prostate cancer receive AR-targeted therapies for prolonged periods, usually until the disease relapses [59]. Therefore, we perturbed all patient-specific networks to mimic prolonged AR-targeted therapy for each patient and compared the percent change in AUC of all nodes before and after perturbation between races, in which AUC represents the temporal activity of a node over an arbitrary unit of time. Simulated AR inhibition resulted in significant differences in the changes in AUC of nodes involved in the TGF-β, IDH1, AR, and cell cycle pathways in prostate tumors between AA and EA men (Additional File 2, Figs. S6A–B). A significantly greater increase in AUC profiles was observed for IDH1 (34.2 vs. 9.6, P < 0.05, Fig. 3A, C) and SMAD (48.5 vs. 15.3, P < 0.05, Additional File 2, Fig. S6A, C, left panels) after AR inhibition in prostate tumors from AA men compared to EA men. Conversely, a significantly greater decrease in AUC profiles was observed for AR_ERG (− 97.4 vs. − 93.4, P < 0.05) and ZBTB17 (− 95.8 vs. − 78.5, P < 0.05) in AA men (Additional File 1, Table S5). The changes in the AUC of all nodes in this prostate cancer network are represented in Additional File 2, Fig. S6A. These results highlight that there are racial differences in the changes in certain gene activities after prolonged AR inhibition. We validated our findings by simulating AR inhibition with The Cancer Genome Atlas (TCGA) [60] cohort. These simulations confirmed race-specific differences in TGF-β, IDH1, and cell cycle pathways (P < 0.05, Fig. 3B, D; Additional File 2, Fig. S6B, D right panels; Additional File 1, Table S5). Overall, our simulations suggest that prolonged AR inhibition can result in changes in gene activity that is significantly different between prostate tumors from AA men and EA men. These observations have real-world implications and need to be investigated in the future since AR inhibition is routinely used to treat advanced prostate cancer.
Fig. 3
IDH1 gene activities after AR knockout simulations show significant race-specific differences in prostate tumors. AUC before inhibition (WT), AUC after AR inhibition (KO), and the percent change in AUC between the two conditions in the Nutrient pathway are represented in the heatmap. A Roswell Park (RP) clinical samples, B TCGA clinical samples. C–D Percent change in IDH1 AUC (highlighted in red) was significantly different between prostate tumors from AA and EA men in both C RP and D TCGA clinical samples. Changes in AUC are represented by blue (down) and red (up)
Metabolic, oncogenic, and immune pathways are dysregulated in prostate tumors from AA menOur prostate cancer Boolean network analyses are limited to a subset of curated genes [15]. Broadening this analysis to encompass all gene expression changes allowed us to pinpoint other race-specific differences in prostate cancer transcriptomes. For this purpose, we compared mRNA levels, obtained from RNA sequencing, between prostate tumors of AA and EA men (AA: T = 31 and EA: T = 32, demographics in Additional File 1, Table S2, samples that were also analyzed by DNA methylation). Genes in peptidase activity pathways, including SEMG1 and SEMG2, were significantly upregulated in prostate cancer from AA men (Fig. 4A, left panel, Additional File 5). To identify race-specific pathway alterations, we performed GSEA using the whole transcriptome data. GSEA revealed peptidase activity, reproductive processes, and microtubule-based movement were overrepresented in prostate tumors from AA men (Fig. 4A, middle panel). Additionally, immune-based pathways that include genes crucial for the proliferation of lymphoid and T-cell progenitors, such as CD40 and CD226, were enriched in prostate tumors from AA men (Fig. 4A, middle panel). The results from the Roswell Park cohort are further supported by the analysis we conducted on the larger TCGA cohort (n = 43 AA; n = 270 EA), which confirmed enrichment of these pathways in AA prostate tumors (Additional File 2, Fig. S7A).
Fig. 4
Race comparisons reveal distinct gene enrichments that correlate with progression-free survival. Left panels represent differentially expressed genes utilizing DESeq2, middle panels represent gene set enrichment analysis based on clusterProfiler, and right panels represent hazard ratios based on calculated GSVA scores. A Comparison of all prostate tumors from AA and EA men. B Race-specific comparisons show distinct pathways in prostate tumors of AA and EA men younger than 55. C Comparisons show distinct pathways in prostate tumors of AA men younger than 55 compared to AA men ≥ 55. NES: Normalized Enrichment Score. An asterisk indicates a significant (p < 0.05) enrichment of the indicated pathway
Gene expression panels based on measuring expression of multiple genes on one panel, such as Decipher [61], can be utilized to derive an overall score for calculating the risk of prostate cancer progression. Therefore, we looked at gene expression in each individual pathway as a potential candidate for ‘gene marker panels’ and calculated GSVA scores. GSVA estimates variation of a gene set and calculates sample-wise (in this case for tumors from individual patients) scores as a function of genes within the given GSEA pathway [42]. While GSEA reveals the enriched pathway, GSVA can be thought of as “activity score” for the genes included in the enriched pathways. Therefore, we calculated GSVA scores for the top pathways identified using GSEA. Using the GSVA scores we determined hazard ratios (HR) based on progression-free survival. Positive HR scores indicate a higher risk of disease progression, and negative HR scores indicate a reduced risk of disease progression. Higher GSVA scores in the microtubule-based movement pathway correlated with positive HR scores and therefore higher risk of disease progression in both AA and EA men (P < 0.05) (Fig. 4A, right panel; Additional File 2, Fig. S7B; Additional File 1, Table S6). The microtubule-based movement pathway included multiple members of the kinesin family that are known to promote disease progression in other cancers [62]. These results lay the groundwork to further develop and test gene expression of the microtubule-based movement pathway in primary prostate cancer and its association with disease progression.
AA men are more frequently diagnosed with early-onset prostate cancer (< 55 years of age) than EA men [63]. To determine race-specific gene expression changes associated with early-onset disease, we identified differentially expressed genes (DEGs) in prostate cancer from < 55 years and ≥ 55 years AA and EA men (Additional File 2, Fig. S7C). Components of the ribonucleoprotein (RN7SK genes) and Y RNA, part of long noncoding RNAs, were significantly upregulated in prostate cancer from AA men < 55 years compared to EA men < 55 years (Fig. 4B, left panel). GSEA revealed an overrepresentation of genes that include enzymes involved in amino-acid metabolism, oxidation of organic compounds, and oxidative phosphorylation (Appierto response in Fig. 4B, middle panel). We again calculated HRs based on progression-free survival and associated GSVA scores to determine whether these pathways correlate with the risk of disease progression. Downregulation of the defense response to bacterium and immunoglobulin production pathways was associated with a significantly lower risk of disease progression only in younger AA men (P < 0.05, Fig. 4B, right panel; Additional File 2, Fig. S7B). Overtreatment of men with low-risk primary prostate cancer remains a significant issue in the clinic [64]. Therefore, our finding represents a potential path forward for identifying AA men with primary prostate cancer with lower risk of disease progression by measuring gene expression changes in the bacterium and immunoglobulin pathways.
To determine tumor-specific gene expression changes, we identified DEGs in prostate cancer from men < 55 years of age and men ≥ 55 years of age by comparing tumors and adjacent non-tumor tissue within each race. RN7SL5P, a long noncoding RNA, as well as other genes, was significantly upregulated in prostate cancer from AA men < 55 years compared to AA men ≥ 55 years (Fig. 4C, left panel; Additional File 5). GSEA revealed that early-onset prostate cancer in AA men was enriched for ribosomal proteins, translational, and metabolism-associated pathways (Fig. 4C, middle panel; Additional File 1, Table S6). Lower GSVA scores of the TMPRSS2-FUSION pathway were associated with a higher risk of disease progression only in AA men < 55 years of age but not in AA men ≥ 55 years of age (Fig. 4C, right panel; Additional File 2, Fig. S7B; Additional File 1, Table S6). Interestingly, we did not identify any significant DEGs or pathways associated with disease recurrence upon comparisons of EA men < 55 years of age and EA men ≥ 55 years of age (Additional File 2, Fig. S7D), suggesting that individual gene expression is similar between these two cohorts. Overall, we identify distinct transcriptional changes associated with the risk of disease progression in AA and EA men with prostate cancer. Future studies of these signaling pathways in preclinical models should pinpoint their mechanistic contributions to prostate cancer biology and disease progression.
留言 (0)