
記住我
The results section is organized based on the following logic. Firstly, we present the obtained association significance data and phenotype description data for three different case studies, visualizing the distribution consistency required for data fusion. Secondly, we evaluate the data fusion results of the PheSeq model, including its performance on the reference dataset DISEASES [39] and a quantitative comparison analysis with results obtained from a single sequence analysis method. Subsequently, we provide an overall comparative observation of the predictions of PheSeq and a single sequence analysis method. Following that, we analyze the positive impact imposed by phenotype description in the PheSeq model. Subsequently, considering that PheSeq incorporates prior knowledge from the literature, we design ablation study to assess PheSeq’s predictive capability by removing prior knowledge. Simultaneously, we conduct a horizontal comparison between PheSeq and several other data fusion methods, comparing the differences in data modalities and data integration strategies. Finally, we develop a phenotype description network to exemplify and showcase the results.
Data visualization for association significance and phenotype descriptionIn the context of three distinct case studies, a total of 24,440 AD-related literature, 55,638 BC-related literature, and 81,463 LC-related literature are fed into the phenotypic embedding generation pipeline. This yields 18,157 gene-AD pairs, 17,374 gene-BC pairs, and 24,578 gene-LC pairs, respectively. We visually represent these associations within the cubic grid in the graphical presentation in Fig. 3. Leveraging the inherent principles of semantic computation, gene-disease pairs with similar phenotypic descriptions are anticipated to exhibit proximity within this embedding space.
Fig. 3
View of data congruence in three case studies. a 3-D semantic representation of AD genes; b BC genes with 3-D representation; c LC genes with 3-D representation. With the color gradient representing the significance level by a single sequence analysis, genes after the phenotypic embedding computation are projected onto a 3-D semantic space. Intuitively, the significant and less significant disease-associated genes are distinguished along the manifold direction based on their phenotypic embeddings. The observation suggests the high data quality of association significance and phenotype description, which supports the subsequent data fusion
To observe the data congruence of the phenotypic embedding and p-value from sequence analysis, we employ a color-coding scheme to visualize the congruence in the distribution of two distinct modalities of data. Here, each gene is colored in a gradient ranging from red to blue, with color intensity denotes the level of statistical significance associated with the p-value of the corresponding gene.
In this figure, the congruence in data distribution between association significance and phenotype description is readily discernible through distinct data partitioning and segmentation. Specifically, genes exhibiting significant p-values (depicted as red dots) tend to disperse across the outer regions of the 3-D manifold space along the manifold path. Conversely, genes with non-significant p-values (represented by green-blue dots) manifest discernible partitioning and segmentation along a distinct direction within the manifold space.
In summary, the significance of p-values aligns with the clustering trend observed in phenotypic embedding. This suggests the potential and rationale for merging embedding data with significant p-values to prioritize disease-related genes. Furthermore, this fusion-based approach has the potential to deepen our understanding of gene-disease associations with the aid of phenotype descriptions.
Evaluation of the predicted genes by PheSeqAfter feeding the association significance data and phenotype description data into the PheSeq model in AD, BC, and LC cases, model iterations ran data fusion processes and generated new p-values for gene-disease associations. Upon the generation of the association significance for each gene after PheSeq implementation, abundant novel gene-disease associations were subsequently suggested.
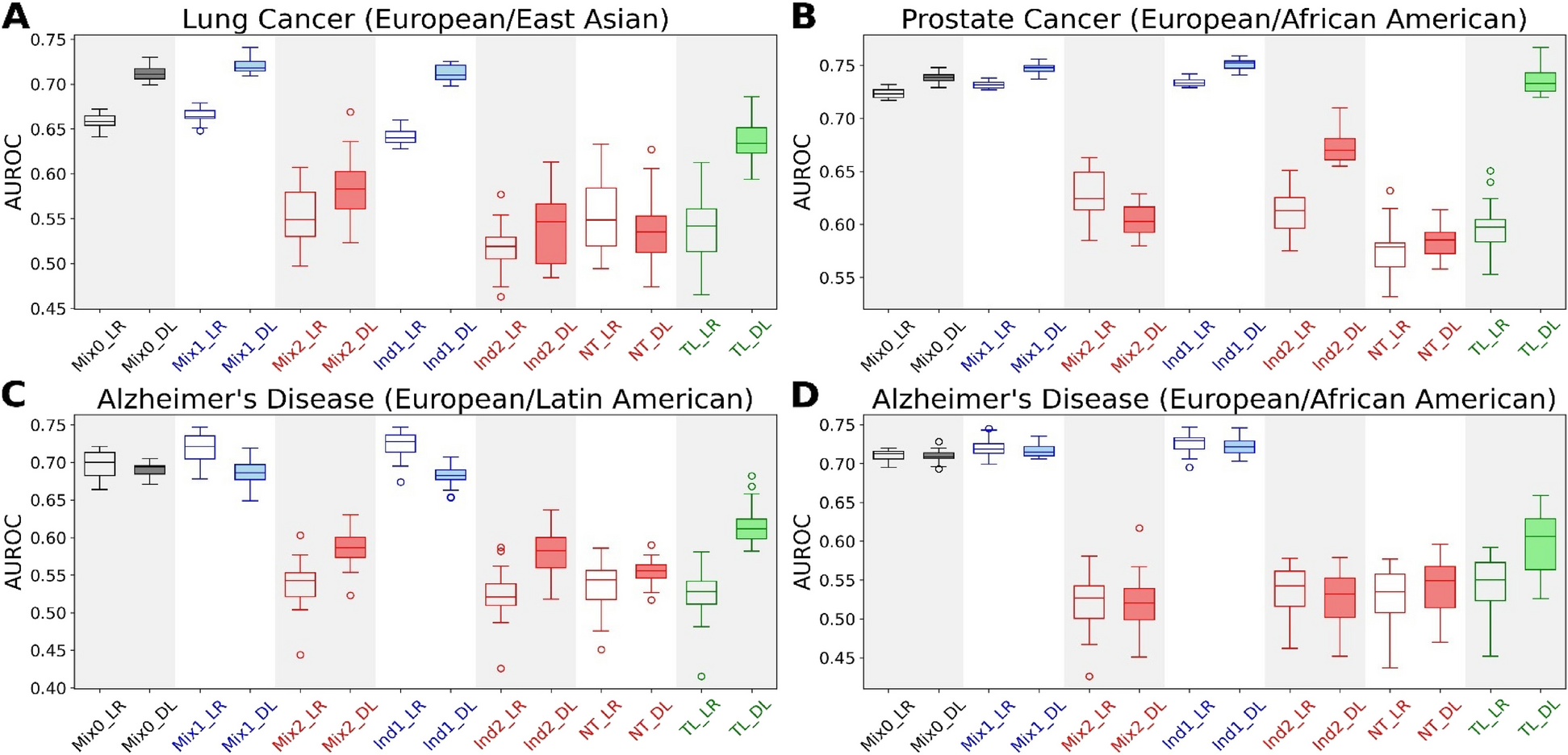
Comparison of prioritized genes by PheSeq and sequence analysis methodsAs illustrated in Table 2, the number of genes predicted by the PheSeq model for AD is 1024. This accounts for 5.6% of the 18,157 background GWAS genes, thereby establishing a moderate ratio when compared to the low positive rate of 1.7% obtained from the GWAS experiment. Similarly, the PheSeq model prioritizes 818 BC genes with a positive rate of 4.7%, which is comparatively higher than the positive rate of 2.7% obtained from the transcriptome experiment utilizing AgilentG4502A_07_3. Furthermore, 566 genes are prioritized for LC, and the resulting positive rate of 2.3% is significantly higher than the positive rate of 0.75% in the methylation experiment with Human Methylation 450.
Table 2 Gene-disease association discovery in the case studies via PheSeq and sequence analysisPheSeq yields newly predicted genes for the association study, of which, a good portion are overlapped ones with single sequence analysis, and the rest are newly recalled ones. In summary, for AD, 236 genes out of 1024 prioritized genes overlap with the GWAS experiment. Similarly, 68 out of 566 genes in LC and 347 out of 818 genes in BC overlap with the methylation experiment and transcriptome experiment, respectively. Furthermore, PheSeq recalls 768, 471, and 498 novel significant genes for AD, BC, and LC, respectively.
Evaluation by the benchmark datasetTo evaluate the prioritization result, a benchmark database, DISEASES [39], is referenced, which integrates 36,448 gene-disease associations from three resources with increasing reliability, i.e., “Text mining,” “Experiments,” and “Knowledge.” Among them, the “Text mining” results are retrieved by text co-occurrence, and “Experiments” collect GWAS databases like target illumination GWAS analytics (TIGA), Catalogue of Somatic Mutations in Cancer (COSMIC), and DistiLD, while the “Knowledge” results involve general gene-disease association databases like AmyCo, MedlinePlus, and UniProtKB.
In AD, three mutually overlapping sources in DISEASES, Text mining, Experiment, and Knowledge, encompass 315, 339, and 26 significant genes, respectively, corresponding to 624 AD genes in total. Among these, PheSeq achieves 128, 48, and 17 gene hits in the three sources, respectively, contributing to 151 significant AD genes in total. The result reveals a relatively higher recall rate, i.e., 17/26, in the Knowledge source, lower in the Experiment source (48/339), and intermediate in the Text mining source (128/315).
The recall rate in BC and LC is higher than that in AD. In BC, the three sources contain 176, 344, and 29 LC genes, contributing to 533 LC genes. The recall rate for each source is 72/176, 73/344, and 19/29. The overall recall rate is 159/533, whereas in LC, the overall recall rate is 342/669. PheSeq hit 119 out of 297 genes in the Text mining source, 239 out of 391 genes in the Experiment source, and 21 out of 25 genes in the Knowledge source. In brief, though it still misses many hits in DISEASES, PheSeq obtains a reasonable recall rate.
By observing the Top 50 genes prioritized by PheSeq (Fig. 4), it is concluded that over half of them have been recorded in the DISEASES database, of which 26/50 for AD, 33/50 for BC, and 36/50 for LC. The high coverage reveals that PheSeq well replicates the associations in the benchmark database. In comparison, the record of significant genes by sequence analysis is relatively scarce within the top 50. It is noted that DISEASES covers a good portion of the significant genes by sequence analysis. Among 311 significant AD genes through sequence analysis, 233 are encompassed in the DISEASES database. In the cases of sequence analysis experiments for BC and LC, 424 out of 470 significant BC genes and 137 out of 184 significant LC genes are cataloged in the DISEASES database.
Fig. 4
The top prioritized genes from PheSeq and sequence analysis in the DISEASES database. The hits plot and cumulative charts in DISEASES serve to compare the recall rate of PheSeq and sequence analysis methods
Interestingly, PheSeq replicates the associations in the three DISEASES resources with different coverage rates. Among them, 24 genes in the AD prediction have been recorded in the “Text mining” part, while the number for BC and LC is 20 and 10. Comparatively, LC obtains higher recall in the “Experiments” part. The results also suggest that it is hard for a single source of data to recover gene-disease associations, and PheSeq is capable of fusing the heterogeneous data to achieve better data comprehension.
Investigation of top prioritized AD-associated genesTaking AD as an example, the top 5 genes with and without GWAS supports are investigated. Being the Top 5, MAPT, PSEN1, APP, APOE, and GRN are known vital ones in the AD pathological hypothesis. In detail, MAPT encodes the Tau protein, and its hyper-phosphorylation forms the neurogenic fiber tangles in neurons and leads to neuronal apoptosis [63]. Moreover, mutations in PSEN1 and PSEN2 have an impact on an APP-cleaving enzyme, \(\gamma\)-secretase, thus regulating APP expression. Meanwhile, the accumulation of Abeta, an APP-encoded protein, forms the fibrillar amyloid plaques in the brain and impairs the ability of spatial learning and memory, which is a known direct cause of AD [64]. Being the most widely studied AD-associated gene, APOE is known to cause neuro-inflammation among AD patients by affecting the microglia [65]. In addition, GRN is a causal gene for frontotemporal dementia, a neurodegenerative disease [66].
Investigation of the top 50 genes leads to a discovery that a good trade-off leverages association significance and phenotype description and helps to infer the potential associations. First, 24 out of 50 prioritized PheSeq genes pass the IGAP GWAS significant test. They both carry significant p-values and supportive semantic evidence. For example, TREM106B, with a significant p-value of 9.53E−14, wins 50 hits in the literature. In addition, 18 out of 50 prioritized genes pass the significant test in the GWAS dataset EFO_0000249. Among them are GRN, TMEM106B, SPI1, CR1, and PICALM from GCST90044699; SORL1 and SQSTM1 from GCST002245; CLU and ABCA7 from GCST90012877; MAPT from GCST90038452; APP from GCST012182; APOE from GCST009019; TREM2 from GCST005549; TTR from GCST007319; FUS from GCST007320; TOMM40 from GCST000682; BIN1 from GCST005922; and ACE from GCST90013835.
Second, for the rest of the 32 genes that do not exist in the GWAS Catalog, 4 of them are included in the known database. In detail, PSEN1 and PSEN2 are both in UniProtKB and MedlinePlus, while SNCA and CSK3B are in UniProtKB. Eventually, for the 28 genes that are not reported by GWAS or known databases, 23 of them are suggested to be AD-related with confirmed phenotype description.
To compare the global prioritization results between PheSeq and sequence analysis, a cumulative chart for database hits for the top 50 filtered genes is given in Fig. 5b.
Fig. 5
The \(-\log p\) plots of overlapping and recalled genes after applying PheSeq and sequence analysis in AD, BC, and LC. a Layout of the \(-\log p\) plot. The x-axis and y-axis denote the \(-\log\) p value from the sequence analysis and the PheSeq model respectively. The red line refers to a strict threshold line such as Benjamini FDR in our case, and the green line refers to a less strict threshold line such as \(-\log 0.005\) in our case. Genes are labeled when overlapped in PheSeq and sequence analysis or recalled by PheSeq. b The \(-\log p\) plot of significance for both PheSeq and sequence analysis in AD. Five genes are marked in red, i.e., MAPT, PSEN1, C9orf72, SOD1, and PSEN2. All of them are PheSeq recalled genes, which obtain high significance in PheSeq but obtain less or limited significance in GWAS. c The \(-\log p\) plot in BC. Five PheSeq recalled genes are chosen and marked in red, i.e., NEU1, ZAP70, EIF2S2, ZNRF3, and CLIC11. These genes obtain comparatively higher significance in PheSeq than that in sequence analysis. d The \(-\log p\) plot in LC. The five marked genes are UGT2B15, VPS33B, ATAD5, GNAT2, and SPPL3. All five genes show strong significance in PheSeq but limited significance in sequence analysis
Overall, the results suggest that the PheSeq model effectively leverages the synergy among heterogeneous association data, alleviating the limitations of using single-source association significance data.
Genes with significance in PheSeq and single-omics sequence analysis: a comparative observationIt is noted that the sequence analysis used in comparative experiments may introduce errors, particularly when considering the inherent instability of results obtained from single-omics sequence analyses. Consequently, the sequence analysis unavoidably overlooks certain known significant associations and may erroneously produce false positive results. PheSeq, on the other hand, aims to reduce the error by data fusion. Therefore, a comparative analysis is performed for both types of experiments.
To display and further investigate each overlapping and recalled gene by considering its significance value both on PheSeq and the sequence analysis, \(-\log p\) plots for all significant genes in AD, BC, and LC are given in Fig. 5. In this figure, the horizontal \(-\log p\) axis refers to the association significance obtained by sequence analysis, while the vertical \(-\log p\) axis corresponds to the generated p-value by PheSeq. In addition, the size of the circle for each gene reflects the count of the phenotype descriptions related to the gene (Fig. 5a).
Intuitively, these figures offer a means by which to investigate the genes with overlapped significance both in sequence analysis and phenotype description. In particular, the plot is separated into sections by threshold lines. The genes with overlapped significance genes are located in the top right corner of the plot, which pass the significance test in sequence analysis and in the meantime carry sufficient association semantics. Meanwhile, the newly reported significant genes by PheSeq are located in the top left section of the plot, which may show less or limited significance in sequence analysis.
In the context of AD, genes with overlapping associations, including APOE, GRN, LRRK2, and SPI1, are visually presented in the top right corner (Fig. 5b), all of which pass the significance test in GWAS and possess sufficient AD-relevance association semantic by PheSeq. Furthermore, genes with less significance in the sequence analysis, e.g., PSEN1, SOD1, MAPT, C9orf72, and PSEN2, are displayed in the top left section. Among the four, PSEN1 and PSEN2 are known AD-related genes, reported in AlzGene [67], while C9orf72 and SOD1 are known to be relevant to neurogenetic disease and possess AD-relevant literature support in GeneCards [68].
In BC, overlapped genes, such as SFRP1, HOXA4, and OSR1 genes, are clearly displayed (Fig. 5c). The location of these genes in the figures show that these genes possess both significance in sequence analysis and PheSeq. Again, we are focusing on the recalled genes by PheSeq. Here, PheSeq recalled less significant genes in sequence analysis such as NEU1, ZAP70, EIF2S2, ZNRF3, and CLIC1, while all of which possess strong significance in PheSeq. The literature review as well shows the relevance of BC to these genes, e.g., NEU1 [69], ZAP70 [70], EIF2S2 [71], ZNRF3 [72], and CLIC1 [73].
Similar observations are carried on for LC, where overlapped genes such as MIR6129, OVAAL, MTOR-AS1, and LINC0269 are displayed in the top right corner of Fig. 5d. Meanwhile, the top left part of the figure indicates PheSeq-recalled genes. The literature review again shows the relevance of LC to these genes, e.g., UGT2B15 [74], VPS33B [75], ATAD5 [76], GNAT2 [68], and SPPL3 [77].
In conclusion, association study results obtained by PheSeq and a single sequence analysis can be simultaneously observed by the above figures. In addition, this figure enables specific investigation on overlapped genes or newly recalled genes by PheSeq after data fusion. The analysis in the three cases suggests that the genes recalled by Pheseq may not be directly associated with the target disease, but they with a high chance exhibit relevance via database or literature review.
Impact of phenotype description on PheSeq with association interpretabilityPheSeq incorporates rich semantic information within its data fusion framework, and it leverages the synergy between the sequence analysis and association descriptions. As a result, PheSeq retrieves a vast dataset of phenotype sentences and bio-concepts for interpreting the prioritized gene-disease association. In summary, 14,084 phenotype sentences are utilized by PheSeq to support 1024 prioritized genes in AD. With an average of 13 phenotype sentences per gene, this dataset includes 1849 GO terms and 1351 HPO terms. In BC and LC, 2250 and 10,440 phenotype sentences are obtained, respectively, with each gene associated with an average of 9 and 10 phenotype sentences. More details on the validation of embedding quality, and the statistic of phenotype description are provided in Additional file 3.
Actually, the PheSeq model prioritizes the gene-disease associations by perceiving corresponding description descriptions. As per observation, genes recalled by PheSeq generally possess pertinent phenotype descriptions. Taking MAPT in AD as an example, it is known to be relevant to the etiology of AD by the widely accepted Tau protein hypothesis, although it fails to pass the significance test in GWAS. As can be observed from Table 3, the most frequently cited phenotype descriptions related to MAPT include “Neurofibrillary tangles” (HP:0002185), “Hyperphosphorylation” (GO:0048151), “Cognitive impairment” (0100543), “Microtubule binding” (GO:0008017), “Long-term synaptic potentiation” (GO:0060291), and “Microtubule polymerization potentiation” (GO:0046784). According to the Tau protein hypothesis, hyperphosphorylation of the Tau protein leads to its aggregation, ultimately disrupting microtubule stability and resulting in the formation of neurofibrillary tangles―a hallmark pathological feature of AD. The observation shows that the top ranked associated phenotype descriptions are highly relevant and supportive for the MAPT-AD association.
Table 3 Associated phenotypes for PheSeq recalled genesWe further investigated four such genes, namely PSEN1, c9orf72, SOD1, and PSEN2, all of which displayed robust significance in PheSeq, despite exhibiting less or limited significance in sequence analysis.
Table 3 presents examples and statistics of the phenotype descriptions including bio-concepts and sentences. Except for C9orf72, the rest of them are all recalled ones by PheSeq. Here, frequently mentioned bio-concepts include “Senile plaques” (HP:0100256), “Neurofibrillary tangles” (HP:0002185), “Hippocampal atrophy” (HP:0410170), “Abnormality of mitochondrial metabolism” (HP:0003287), and “Inflammatory response” (GO: 0006954). These phenotype descriptions are known to be relevant to AD, thus suggesting a potential gene list for further AD-gene association investigations.
Similarly, an inquiry is undertaken regarding NEU1, ZAP70, EIF2S2, ZNRF3, and CLIC1 in BC. Remarkably, these genes exhibit significant importance in PheSeq analysis, despite showing relatively modest significance in sequence analysis.
In accordance with the aforementioned observations in the AD case, phenotype descriptions with high association relevance are derived. Specifically, bio-concepts such as “Angiogenesis” (GO:00001525), “Cytokine production” (GO:0001816), “Epidermal growth factor-activated receptor activity” (GO:0005006), “Aldehyde dehydrogenase [NAD(P)+] activity” (GO:0004030), and “Wnt signaling pathway” (GO:0016055) are frequently mentioned.
Meanwhile, UGT2B15, VPS33B, ATAD5, GNAT2, and SPPL3 exhibit a significant impact on PheSeq in LC and win corresponding literature support [68, 74,75,76,77], despite not meeting the reference threshold in sequence analysis. Consistent with previous observations in AD and BC cases, these genes are commonly associated with LC-relevant phenotypes, including “Low-density lipoprotein particle receptor activity” (GO:0005041), “Fibroblast growth factor-activated receptor activity” (GO:0005007), “GDP-dissociation inhibitor activity” (GO:0005092), “Goodpasture-antigen-binding protein kinase activity” (GO:0033868), and “Transforming growth factor beta receptor binding” (GO:0005160).
In summary, these results indicate that PheSeq underscores the disease-specific phenotype descriptions and incorporate them with sequence analysis significance. Remarkably, PheSeq holds particular importance in situations where a single sequence analysis may elicit systematic bias and flawed predictions of crucial genes. In such instances, PheSeq serves as an effective tool for establishing a connection between phenotype descriptions and association significance in sequence analysis and helps to recall the significant genes.
Impact of prior knowledge on PheSeq with association prediction: an ablation studyIn the aforementioned analysis, we compare the performance of PheSeq with that of a single sequence analysis in three distinct case studies. It is essential to note that, as a data fusion method, PheSeq inherently incorporates prior knowledge from literature and networks. Consequently, PheSeq is a model integrating prior knowledge and holds an inherent advantage over conventional sequence analysis models. In this section, we conduct an ablation study to evaluate how prior knowledge is incorporated into the PheSeq model. We systematically remove specific prior information and rerun the entire prediction process to assess the impact accordingly.
Based on the publication dates of omics data, we exclude all literature data beyond those time points. Specifically, for AD, the literature cutoff date is set at October 27, 2013. Correspondingly, for BC and LC, the respective dates are January 28, 2016. Consequently, this approach results in a significant compression of the prior knowledge derived from the literature. In the original experiments, the literature on AD covers 14,261 genes; however, with the cutoff set on October 27, 2013, only 1017 genes are now covered. In the case of BC, the gene coverage decreases from 10,498 to 3,399, and in LC, the reduction rate is greater, dropping from 20,460 to 749 genes.
PheSeq in the ablation setting predicts 391 significant genes associated with AD, 1398 significant genes associated with BC, and 172 ones with LC. Despite the relatively limited inclusion of prior literature knowledge for these genes, the results in Table 4 clearly demonstrate two patterns. First, predicted significant genes typically carry a higher proportion of literature knowledge. For instance, among the 391 key AD genes, each gene, on average, possesses 21.17 literature references, 31.80 pieces of related sentence evidence, and 11.54 core concepts, whereas in the corresponding non-significant genes, these values are only 2.32, 3.28, and 2.90, respectively. Second, due to the preservation of PPI data in prior knowledge, prioritized genes are more likely to be adjacent to other significant ones. For instance, among the 391 AD significant genes, statistical analysis of information from their top 10 neighbors reveals an average of 5.54 significant genes per gene, with a cumulative literature count of 115.35, a sentence evidence count of 173.22, and a concept count of 62.86. In contrast, for non-significant genes, the number of significant genes among their top 10 neighbors decreases to 3.13, with corresponding literature, sentence, and concept counts of 3.08, 4.36, and 3.87, respectively. The two patterns are observed as well in BC and LC case studies.
Table 4 Investigation of prior knowledge derived in the significant or non-significant genes in the ablation studyIn short, significant genes exhibit extensive prior knowledge, either encompassing abundant literature in historical data or demonstrating strong associations with significant disease-related genes in PPI networks.
Taking PICALM as an example, this gene is notably associated with a substantial amount of AD literature. As of the end of 2023, a total of 264 publications are available for PICALM, with 112 publications retained before the cutoff in 2013. This abundance of literature contributes to PICALM being identified as a significant gene with a high probability in the ablation study conducted by PheSeq. Similarly, ESR1 in LC also maintains a considerable literature count, totaling 132 publications by the end of 2023 and retaining 54 publications before the cutoff in the preceding years of 2016.
In AD, GBA emerges as the gene exhibiting the strongest association in the PPI network. Its neighbors, such as UGCG, PSAP, GALC, and SGMS2, are all linked to known AD pathological processes and exhibit significant p-values in sequence analysis, namely 0.045, 0.029, 0.00056, and 0.0014, respectively. This significantly increases the likelihood of PheSeq identifying GBA as a significant gene.
Similarly, in BC, the NEU1 gene is strongly linked to several significant genes in the PPI network, including GLB1 (4.11e−11), ARSA (5.15e−05), and GAL3ST1 (1.99e−13). This, in turn, leads to PheSeq maintaining positive predictions for these genes in the ablation study.
In summary, the observed patterns in the ablation experiments indicate that despite the extensive removal of literature prior knowledge, the predicted significant genes still predominantly retain both literature and network priors. This in turn aligns with the initial purpose of data fusion.
Furthermore, we evaluate the predictive capacity of PheSeq with removed prior literature knowledge, and the top 50 significant genes with the cumulative charts in DISEASES are shown in Fig. 6a. The yellow line represents the ablation method where literature priors are excluded, while the red line corresponds to the original PheSeq method. In the cumulative line plot, it is observable that the yellow line consistently remains below the red line. This result indicates a significant decline in the predictive capability of PheSeq when a substantial amount of literature priors is removed, and it aligns with the data fusion concept in PheSeq.
Fig. 6
The top 50 significant genes with cumulative charts in DISEASES in the ablation study
Comparison of other data fusion modelsAs a representative data fusion algorithm, PheSeq combines two distinct types of association information: sequence analysis data and embedding data. When addressing gene-disease associations, there are diverse strategies for data incorporation and model selection within data fusion algorithms. Even when examining the same disease, variations in results among different fusion methods can arise due to the use of diverse data modalities. Figure 7 illustrates the overlap of significant genes under various methodologies. As depicted in the figure, achieving a high degree of overlap between different methods is challenging, regardless of the number of significant genes predicted by each approach.
Fig. 7
Overlap of significant genes from different data fusion methods on gene-disease associations. a AD. b BC. c LC
Nevertheless, conducting a comparative analysis of outcomes from various data fusion methods, including PheSeq, remains crucial for obtaining a comprehensive evaluation of PheSeq’s performance. As shown in Table 5, different methods cover various data modalities, including data from GWAS, gene expression, gene regulatory network (GRN), expression quantitative trait loci (eQTL) high-throughput chromosome conformation capture (Hi-C), copy number alteration (CNA), literature, and protein-protein interaction. The number of significant genes varies across methods, with Lee et al. [78] having the lowest at 12 and PheSeq having the highest at 1024 in AD. In BC, Kim et al. [79] report the lowest count at 35 while PheSeq has the highest count at 818. In LC, Zhang et al. [80] have the lowest at 23 whereas PheSeq exhibits the highest at 566. This likely reflects differences in the identification of significant genes when using different methods and data modalities.
Table 5 Comparison of different data fusion methods on gene-disease associationsThere are three main types of data fusion strategies used in machine learning; early (data-level), intermediate (joint-level), and late (decision-level) [89, 90]. In the early data fusion algorithms, data from various sources, once fully collected, are mapped to a unified data space through vectorization methods such as concatenation or addition. Subsequently, a machine learning model is employed for knowledge-based decision-making. Researches [78, 80, 81, 83,84,85, 88] fall into this scope. In contrast, intermediate data fusion algorithms often utilize a series of models within a step-wise set, where different models handle distinct stages of data, ultimately completing data fusion and knowledge-based decision-making within a single pipeline. This type of algorithm includes researches [79, 82, 86, 87]. Late data fusion algorithms, on the other hand, involve the simultaneous processing of data from different sources by various models, achieving integrated decision-making. Although the selected comparative experiments only represent a small portion of the data fusion methods for three case studies, it is suggested that early and intermediate data fusion methods remain predominant, and late data fusion methods are relatively less frequently employed. GDAMDB [32] and PheSeq stand as representatives of late data fusion methods, utilizing Bayesian networks to learn the distribution relationships among data variables, offering interpretable fusion decisions.
In addition, the interpretation approaches vary widely among these methods. While some methods rely on enrichment analysis and pathway analysis, others incorporate more sophisticated techniques such as interpretable neural networks or recommendation systems. Additionally, some methods do not explicitly specify their interpretation approach. This diversity highlights the complexity of interpreting integrated data and underscores the need for tailored approaches based on the specific objectives of each study.
Finally, we utilize DISEASES as the external dataset referenced to compare the performance of predictive capacity among these methods. As detailed in the rightmost column of the table, PheSeq exhibits superior predictive performance in BC and LC, outperforming other methods in precision and recall. For instance, PheSeq recalls 159 DISEASES genes out of 818 predicted significant genes. Both the amount and the ratio are greater than the rest methods. In AD, while PheSeq recalls 151 DISEASES genes, this is attributed to its larger overall prediction quantity. Conversely, Lee et al. [78] and GDAMDB [32] demonstrate higher precision, with GDAMDB displaying notably high recall values. This also underscores the advantages of the late data fusion approach.
In summary, PheSeq stands out as a late data fusion algorithm in the context of gene-disease associations, predominantly employing phenotype descriptions extracted from literature to enhance the interpretive aspects of the obtained results.
Association interpretation in a visualized phenotype description networkBenefiting from the good amount of phenotype description and sentence support, we derive abundant phenotype descriptions for gene-disease associations. To summarize all the PheSeq-prioritized genes with the collected bio-concepts and sentences, a visualized phenotype description network is built for AD, BC, and LC, separately. In the network, the significant genes (both from PheSeq and sequence analysis) and the bio-concepts are treated as nodes, and a gene-concept edge is linked when a sentence description addressing the association is available. The network is released in a user-friendly webpageFootnote 1, while the pipeline of the network construction is introduced in Additional file 3.
The network offers diverse patterns of association interpretations that serve to enhance the comprehension of the mechanisms that underlie gene-disease associations.
Pattern 1. GO enrichment analysisThe network enables GO enrichment analysis. Here, four gene sets are shown in Fig. 8a, b, c with GO terms corresponding to apoptosis [91], mitophagy [
留言 (0)