
記住我
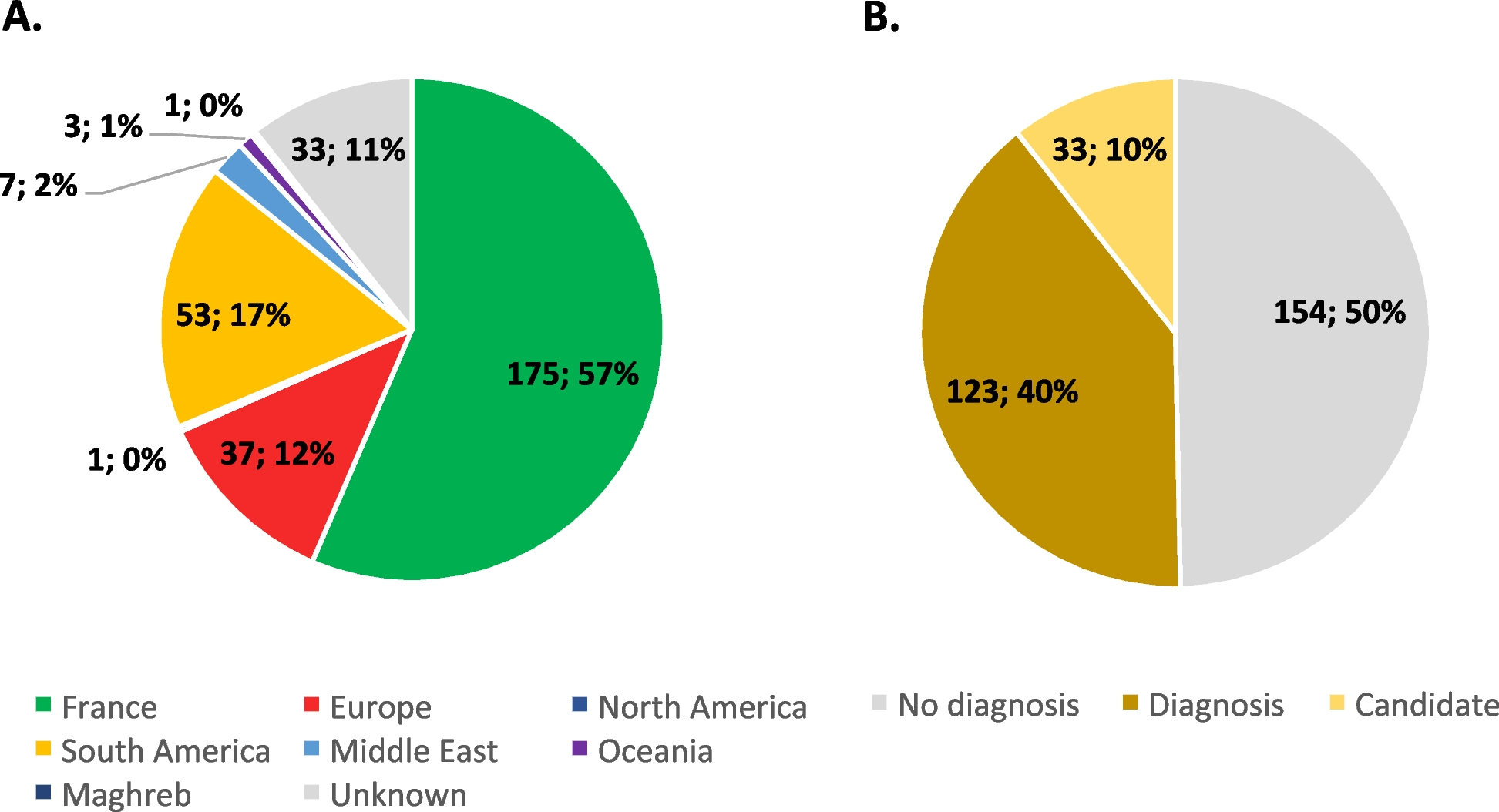
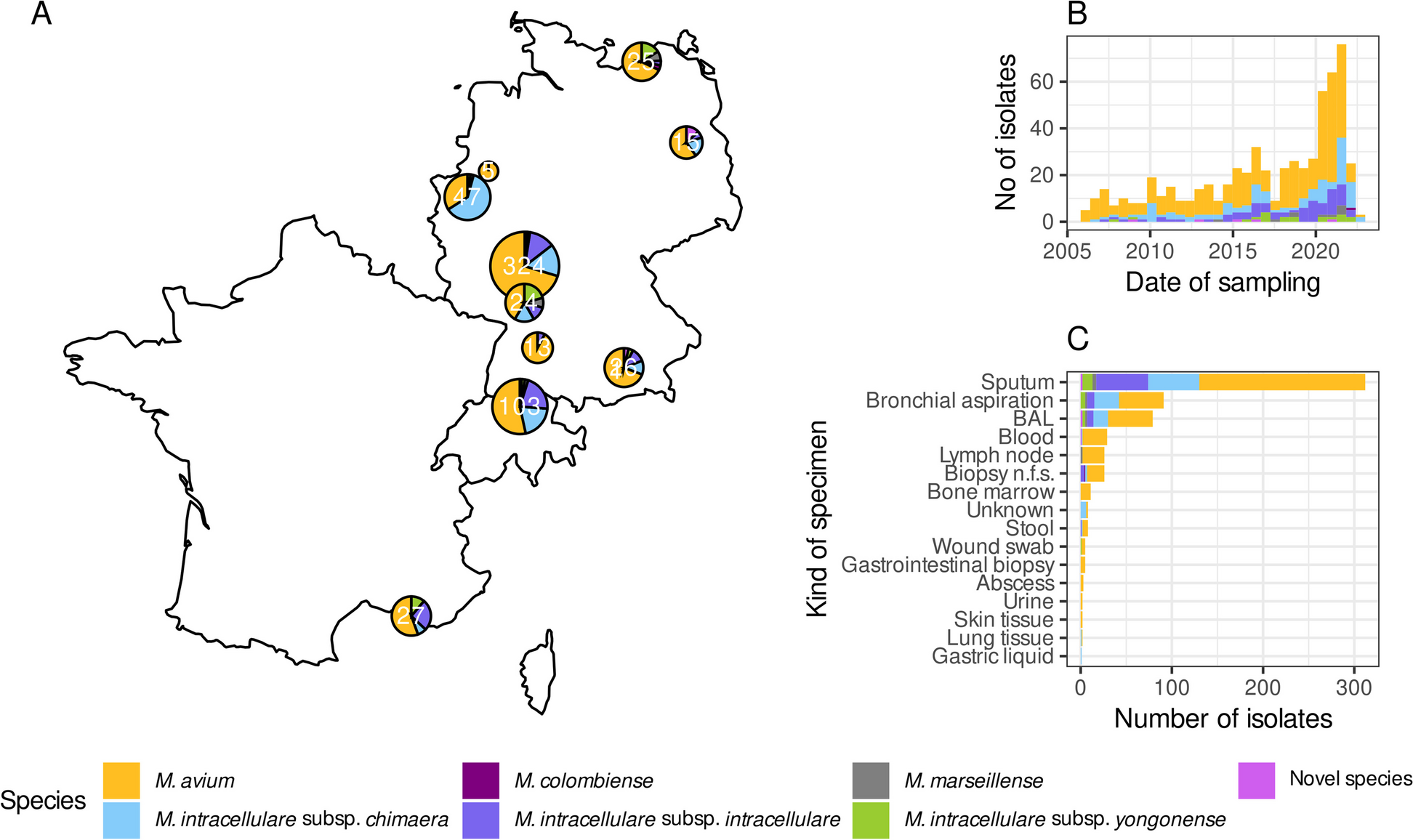

Patient recruitments three cohorts of patients were recruited: 23 scRNA-seq, 365 bulk RNA-seq PitNETs, 128 Immunohistochemistry (IHC) of TMA, and 45 flow cytometry for analysis and validation. All patients received surgery at the Department of Neurosurgery at Ruijin Hospital, an affiliate of Shanghai Jiao Tong University School of Medicine. The scRNA-seq and flow cytometry PitNETs cohort underwent surgery between 2020 and 2023, while the bulk RNA-seq and IHC PitNETs cohorts were collected between 2017 and 2022. Both cohorts only included subjects without previous malignancies. The clinical data of all patients were retrospectively obtained from medical records (Additional file 2: Table S1-5). The patients are assigned numbers matching the GroupsIndex column in Supplementary Table S1-5. Written informed consent was obtained from all patients, and the Ruijin Hospital ethical committee approved the study.
Tissue dissociation and primary tumor cell extractionTissues were transported in DMEM (Gibco, Cat. no. 11875–093) with 1 mM protease inhibitor (Solarbio, Cat. no. P6730) on ice to preserve viability, washed two to three times with phosphate-buffered saline (PBS; Hyclone, Cat. no. SH30256.01), then minced on ice. We used dissociation enzyme cocktail 1 mg/ml Type VIII Collagenase (Sigma-Aldrich, Cat. no. C2139), 2 mg/ml Dispase II (Sigma-Aldrich, Cat. no.4942078001), 1 mg/ml Trypsin Inhibitor (Sigma-Aldrich, Cat. no. T6522), and 1 unit/ml DNase I (NEB, Cat. no. M0303S) dissolved in serum-free DMEM to digest the tissues. Neoplastic tissues were dissociated at 37 °C with a shaking speed of 50 r.p.m for about 40 min. We repeatedly collected the dissociated cells at intervals of 20 min to increase cells yield and viability. Cell suspensions were filtered using a 40-μm nylon cell strainer (Falcon, Cat. no. 352340) and red blood cells (RBC) were removed by RBC lysis buffer (Invitrogen, Cat. no. 1966634) with 1 unit/ml DNase I. Dissociated cells were washed with PBS containing 0.04% bovine serum albumin (BSA; Sigma-Aldrich, Cat. no. B2064) with step-by-step descending centrifuging speed and increasing time. Cells were stained with 0.4% Trypan blue (Invitrogen, Cat. no. T10282) to check the viability and then cultured in DMEM containing 10% FBS and 1% antibiotic mixture.
RNA sequencing alignmentTo quantify gene expression in the transcriptome, the RNA sequencing raw FASTQ files were aligned to the human reference genome GRCh38 (release 40). The human reference genome and its annotation file were acquired from the GENCODE database, accessible at https://www.gencodegenes.org/. Salmon (v1.8.1) [29] was used to generate the count and transcripts per kilobase of exon model per million mapped reads (TPM) matrix. The transcript counts were combined using DESeq2 [30] and then transformed into fragments per kilobase million (FPKM) to normalize the gene length, using the TPM matrix to evaluate the gene expression level. To identify differentially expressed genes, we used the limma [31] package with a significance level set at an adjusted P-value < 0.05 and a log2-transformed fold change (log2FC) > 0.58 (FC > 1.5). To identify differentially expressed genes between more than two conditions, we used analysis of variance (ANOVA) to calculate the P-values.
Functional enrichment analysisGene Set Enrichment Analysis (GSEA) [32] was employed with the pre-ranked algorithm and the R package clusterProfiler [33] to perform functional enrichment analysis. Genes were ranked based on their log2FC. The gene sets from the Molecular Signatures Database (MSigDB, v7.5.1) [34] of the Broad Institute were used. Specifically, we used the HALLMARK gene sets (H) [34], which represent 50 well-defined biological processes, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) [35, 36] database to identify differences induced by various drugs. A significance cutoff of P-value < 0.05 was considered. To visualize the enrichment results, we utilized the enrichplot R package (https://bioconductor.org/packages/enrichplot/). Another functional enrichment analysis algorithm, Enrichr (https://maayanlab.cloud/Enrichr/) [37] web service, was conducted to evaluate gene signatures. P-value < 0.05 was set to the significance level.
Single-cell RNA sequencing alignment and generation of gene expression matrixRaw sequencing reads of the cDNA library was processed through the BD Rhapsody Whole Transcriptome Assay Analysis Pipeline (v1.8), which included filtering by reads quality, annotating reads, annotating molecules, determining putative cells, and generating a single-cell expression matrix. The pipeline also selected the sample origin of every single cell via the sample determination algorithm according to the sequencing reads of the SampleTag library. Among all the output files, a matrix of UMI counts for each gene per cell was used for downstream analysis. Genome Reference Consortium Human Build 38 (GRCh38) was used as a reference for the BD pipeline.
Analysis of single-cell expression data and cell type annotationGene expression matrices were imported for downstream analysis using the R Seurat package [38].
Single-cell quality control (Additional file 2: Table S6)In the initial step, we conducted a filtration process to eliminate cells exhibiting low gene expression. The gene expression matrices of individual cells were imported for subsequent analysis using the R Seurat package. Cells with expressed genes < 200, or unique counts > 50,000 or < 500, or expressed mitochondrial RNA > 30% were removed for quality control, resulting in 98,523 cells being retained. Upon analyzing the cell distributions across the 23 samples, the median number of captured cells was 2837. Among the 23 samples, 6 had a cell count exceeding 5000, with 2 samples notably containing a high number of captured cells (> 10,000 cells). To ensure effective integration, we employed random downsampling to limit the maximum number of enrolled cells for each sample to 5000 using the R sample function. Ultimately, a total of 69,539 cells from PitNETs were included for downstream data analysis (Additional File 2: Table S2). We performed the doublets analysis by using the R package DropletUtils (https://bioconductor.org/packages/DropletUtils/); we identified 108 cells (0.15%) as potential doublets among the total 69,539 cells.
Batch effect adjustmentFollowing the generation of the Seurat object containing 69,539 cells, we identified the top 3000 highly variable genes and utilized them to conduct principal component analysis (PCA). Batch effect correction across samples was executed using the R package Harmony [39], with the parameter “max.iter.harmony” set to 5. Subsequently, the top 20 harmony coordinates were chosen for clustering analysis and dimensionality reduction.
Clustering and dimensionality reductionThe top 20 harmony coordinates were selected for graph-based unsupervised cell clusters with the resolution was set to 0.8. Dimensionality reduction methods t-Distributed Stochastic Neighbor Embedding (t-SNE) [40] and Uniform Manifold Approximation and Projection (UMAP) [41] was performed for visualization. The t-SNE was selected for final interpretation.
Annotation of major cell populationsAfter clustering, 30 clusters were calculated. Marker genes for each cluster were calculated using the “FindAllMarkers” function under the following criteria: log2 fold changes (log2FC) > 0.25, min.pct > 0.1, and adjusted P < 0.05. Cells were annotated using both machine-learning-based software SingleR [42] and high expression of canonical markers (i.e., EPCAM for epithelial cells, NCAM1 for neuron cells, VWF for endothelial cells, DCN for fibroblast. PTPRC for immune cells, C1Q for macrophage/Dendritic-cells (MDC), and CD3 for T cells, CD79A for B cells) in each cluster. For the second step of cell annotation, the immune populations were extracted separately and performed the entire workflow of normalization, batch effect correction, unsupervised cell clustering, and cell annotation and identification. The small cell population inner the immune cell populations were finally identified.
Identification of tumor cells in PitNETsThe tumor cells’ identification was described as (i) expression of neuron markers (NCAM1) and canonical markers of PitNETs, such as POU1F1, TBX19, and NR5A1 and (ii) higher copy number variation level. The copy number karyotyping analysis was performed by inferCNV (https://github.com/broadinstitute/infercnv) and CopyKAT (https://github.com/navinlabcode/copykat) [43] package with default parameter.
Differentially expressed genes calculation and transcription factors activities estimationWe employed Seurat's “FindMarkers” feature to detect genes expressed at varying levels between separate cell types. The log fold change (logFC) threshold was set at 0.01, and the minimum percentage of cells expressing the gene (min.pct) was set at 0.01. The significance of DEGs was determined by log2FC > 0.5 and adjusted P-value < 0.05. Protein activities of transcription factors (TFs) were estimated using DoRothEA [44]. A significance criterion of P < 0.05 was employed to identify variations in TF activity across various cell types.
Generation of cell signatures based on scRNA-seqThe top 200 expressed genes (log2FC > 0.58 and adjusted P-value < 0.05) in each cell type were selected as an in-house cell marker database of PitNETs, which was enrolled for analysis on bulk RNA-seq data. The enrichment score to infer cell abundance from bulk RNA-seq was calculated using a single sample gene set enrichment analysis (ssGSEA) algorithm via the R GSVA [45] package.
Trajectory analysis of macrophagesIn order to assess the possible dynamic changes in cell state across several subclusters of tumor-associated macrophages (TAM), we employed trajectory analysis using the R package Monocle2 (version 2.24.0) [46]. The DDRTree method was used to reduce dimensionality based on the top 50 DEGs in each TAM subcluster and monocyte. Based on the trajectory results, monocytes were defined as the initiating point of the trajectory. Genes with branch-dependent (CX3CR1+ macro and C1Q+ macro) expressions were identified using the BEAM subprogram, filtered with a q-value < 0.05, and visualized by plot_genes_branched_heatmap function. ScTour and DifussionMap were utilized with the default parameters.
Cell–cell communication analysisWe applied the R package Cellchat to evaluate the interaction weights between tumor cells and TAMs [47]. First, we created a cell chat object with default parameters. The ligand–receptor interaction database we used for analysis was “CellChatDB.human” without additional supplement.
NicheNet was applied to infer the interaction mechanisms between TAMs and malignant cells [48]. We defined the niches of interest for every subcluster. Clustered cells with gene expression over 10% were considered for ligand and receptor interactions. The top 100 ligands and top 1000 targets of differentially expressed genes of “senders” and “receivers” were extracted for paired ligand–receptor activity analysis. When evaluating the regulatory network of TAMs on tumor cells, NR5A1 + tumor cells were considered “receivers”.
In contrast, POU1F1+ tumor cells and TBX19+ tumor cells were used as reference cells to check the regulatory potential of TAMs on tumor cells. The ligand_activity_target_heatmap in Nichenet_output was used to show the regulatory activity of ligands. Activity scores ranged from 0 to 1.
Spatial transcriptomics analysisThe stereo-seq data is preprocessed using the STOmics Analysis Workflow (SAW). The read mapping pipeline uses STAR to align the sequenced reads to the human reference genome (GRCh38). Reads for which mass value (Q = − 10log(err rate)) is less than 15 account for more than 40% of the total number of bases, or N reads with more than 5 bases or containing linker sequences were filtered. Valid CID sequence must completely match chip barcode sequence. The gene quantification pipeline uses Bam2Gem to count the number of reads mapped to each gene. Bin200 was set as the basic unit for further data statistics.
The SpatialFeaturePlot function in Seurat generated spatial feature expression plots (Additional file 2: Table S7). Signature scores were integrated into the “metadata” of the spatial transcriptomics (ST) dataset by calculating the mean expression levels of each gene from the scRNA-seq dataset. The estimation of cellular composition for the spatial transcriptomics spots was performed using the SpatialDecon function, and pie plots visualizing the deconvolution results were generated using the DeconPieplot function in the Cottrazm package (version 0.1.1) [49].
Immunohistochemistry staining of tissue microarrayThe tissue microarray (TMA) dataset (n = 128, Additional file 2: Table S3) was used to evaluate the immune cell infiltration. Following deparaffinization and rehydration, heat-induced epitope retrieval (HIER) was performed by submerging the slides in antigen unmasking solution (Solarbio).
After blocking endogenous peroxidase and nonspecific binding sites (0.3% H2O2 and 5% normal goat serum, sequentially), primary antibodies were applied at 4 °C overnight. Slides were incubated with Dako REAL EnVision HRP rabbit/mouse (belong to K5007, DAKO, Glostrup, Denmark) at RT for 20 min, followed by treatment with Dako REAL DAB + CHROMOGEN and Dako REAL substrate buffer (belong to K5007, DAKO, Glostrup, Denmark) to visualize staining signals under light microscopy, finally counterstained using hematoxylin solution. Stained slides were scanned using Ocus (Grundium, Tampere, Finland) and analyzed with Qupath software (see below).
IHC image analysisStained slides were scanned using Ocus (Grundium, Tampere, Finland) and analyzed with Qupath software v0.3.0. The built-in stain vector estimator preprocessed images. Cells with shape and stain parameters in each area were identified by build-in cell detection via nucleus stain (hematoxylin). The threshold for positive mean DAB optical density (OD) was determined based on the staining pattern and intensities observed in all photos for each antibody. The percentage of CD45, CD68, CD8, Ki67, and Cleaved Caspase 3 positive cells were quantified using a customized cellular multiplex algorithm. Scripts were produced for the analysis methodology of all the above slide pictures. These scripts were then executed in batches for each set of images and subsequently reviewed by two pathologists who are experts in the field. All quantifications were evaluated blinded to patient clinical information and outcomes.
Multiplex immunohistochemistry and analysisFormalin-fixed and paraffin-embedded tissue Sects. (3 mm) were de-paraffinized and rehydrated. Next, heat-induced epitope retrieval (HIER) was performed, followed by blocking with 3% hydrogen peroxide in TBST for 10 min and staining with the multiplex mIHC kit (PerkinElmer, NEL861001KT, Shanghai Kelin Institute). Briefly, after the first primary antibody staining, slides were incubated using the HRP-polymer detection system for 10 min, then visualization using Opal TSA working solution (1:100) for another 10 min. Afterward, antigen retrieval was conducted again to prepare the slides for the next antibody. Using this Opal staining method, primary antibodies were applied sequentially. Lastly, slides were counterstained with DAPI (Sigma, 1:1000) for nuclei visualization and subsequently coverslipped using the Hardset mounting media (VectaShield, H-1400).
All tissue sections that underwent multiplex fluorescent staining for each fluorophore were imaged using the Vectra Polaris imaging system (PerkinElmer, Shanghai Kelin Institute) under the appropriate fluorescent filters to produce the spectral library required for multispectral analysis. A whole slide scan of the multiplex tissue sections produced multispectral fluorescent images visualized in Phenochart (PerkinElmer).
Cell culture and reagentsThe GH3, AtT20, MMQ, and RC-4BC cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). GH3, AtT20, and MMQ cell line Twere cultured in Ham's F-12 K medium (L450KJ, BasalMedia) supplemented with 2.5% FBS (S615JY, BasalMedia), 15% horse serum (26,050,088, ThermoFisher), and 1% penicillin/streptomycin (C100C5, NCM biotech. RC-4BC cells were cultured in DMEM (L130KJ, BasalMedia) with 10% FBS, 5 ng/ml recombinant rat EGF (ab290070, Abcam), and 1% penicillin/streptomycin. The following inhibitor and recombinant protein were used: recombinant INHBA (C687, Novoprotein), SB-505124 (HY-13521, MedChemExpress), A 83–01 (HY-10432, MedChemExpress), and recombinant human Follistatin (10685-H08H, Sino Biological).
Flow cytometry and cell sortingFor surface staining, cells were resuspended in 50 μl of PBS containing antibody cocktails and stained at room temperature in the dark for 30 min. Antibodies used for flow cytometry are listed (Additional file 2: Table S8). For intracellular staining, cells were fixed and permeabilized by Foxp3 Fixation/Permeabilization kit (Thermo Fisher Scientific) at 4 °C for 45 min and stained with 50 μl of 1 × permeabilization buffer containing antibody cocktails at 4 °C in the dark for 45 min.
For cell sorting, among the immune cells from human pituitary tumor cell suspensions, CD45-positive were divided into T cells (CD45 + CD3 +), B cells (CD45 + CD3-CD19 +), NK cells (CD45 + CD3-CD19-CD56 +), and macrophages (CD45 + CD3-CD19-CD56-CD11b +); among the CD45-negative, non-immune cells were divided into epithelial cells (CD45-EPCAM + CD31-), endothelial cells (CD45-CD31 + EPCAM-), and stromal cells (CD45-CD31-EPCAM-CD90 +). Among CD14-macrophages, CX3CR1 + , C1Q + , and Lyve + were used to classify macrophages, respectively. The gating strategy is shown in Additional file 1: Fig S11.
All flow data were acquired by BD FACSDiva software v8.0.2 and analyzed by FlowJo VX.
TAM and tumor cell co-culture systemWe isolated CX3CR1+ TAMs from three independent patients (334, 345, and 356) belonging to the SF1 lineage. The clinical details for these patients are provided in Supplementary table s3. We co-cultured the sorted CX3CR1+ TAMs with matched CD45- tumor cells. Briefly, 1 × 104 TAMs were sorted and seeded onto Transwell polycarbonate filters (0.4 µm pore, 6.5 mm membrane diameter; Corning Incorporated, Corning, NY, USA). Simultaneously, 5 × 105 primary tumor cells were seeded into the lower compartments of Transwell chambers, allowing co-culture with TAMs in the upper compartment for 72 h. The initially identified tumor cells from the lower compartment were then collected for further investigation.
CellTiter-Glo luminescence assayCellTiter-Glo luminescence assay (Promega, Madison, WI) was used to determine the suppressive effect of the corresponding drug on the control or gene-edited cell line. The cells were distributed at 2000 cells per well on a 96-well plate. The cells were exposed to the appropriate drugs in a 10% FBS-F12K medium at the prescribed doses. Cells were cultured for the indicated time before adding 100 μL of the CellTiter-Glo® luminescence assay reagent in each well. Cells were incubated for an additional 10 min at room temperature to stabilize luminescent signals and transferred to 96-well black plates. Measurements were performed using a luminescence reader (TECAN, Männedorf, Switzerland). Data was analyzed by GraphPad Prism 5 and normalized to the control group. The p-value was calculated using the Student's t-test. Data was generated from at least three independent experiments.
Cell apoptosisFlow cytometric analysis was conducted using the Annexin V, FITC Apoptosis Detection Kit (AD10, Dojindo) to evaluate cellular apoptosis. Primary tumor cells were harvested and rinsed twice with PBS. Subsequently, the cells were incubated with Annexin V-FITC/PI per the manufacturer’s instructions. The cells were acquired and analyzed using BD FACS Calibur with FlowJo (version 7.6.1) software.
Immunofluorescence and analysisThe cells were derived from samples 375, 381, and 382. Sorted 1 × 104CX3CR1+ and CX3CR1− macros were centrifuged on a slide by Thermo Scientific Shandon Cytospin. After that, cells were washed once with PBS and then fixed with 4% paraformaldehyde for 5 min at room temperature. Cells were washed thrice with PBS and permeabilized with 0.3% Triton-X for 5 min at room temperature. After three times PBS washing, cells were blocked at room temperature with 5% FBS blocking solution for 30 min. The cells were incubated overnight at 4℃ with anti-INHBA and anti-CD68 primary antibodies in a blocking solution. The cells were then washed for 5 min at room temperature three times with PBS. The cells were then incubated with secondary antibodies for 1 h at room temperature.
Anti-goat FITC for INHBA and anti-rabbit Corelite 594 for CD68 were used. All secondary antibodies were diluted in PBS. After that, the cells were washed at room temperature three times with PBS and stained with Hoechst at 1/1000 (40731ES10, yeasen) for 5 min at room temperature. Finally, the slides were washed three times with PBS and mounted on glass slides with AntiFade Mounting Medium ( G1401-5ML, Servicebio). Image acquisition was performed with an Axio Imager M2 (Carl Zeiss Ltd) and an Apotome. The Zen software piloted 2 (363 oil immersion objective) (Carl Zeiss Ltd). Additional file 2: Table S8 summarizes the antibodies used and how they were diluted.
To quantify the fluorescence intensity, we used ImageJ software with default parameters. The grayscale value assigned to each pixel in the single-channel fluorescence image reflects the corresponding fluorescence intensity. The INHBA mean fluorescence intensity (MFI) was determined using the formula: Mean Fluorescence Intensity (MFI) = Total Fluorescence Intensity in the Region / Area of the Region. The grayscale value and the corresponding area were determined for each region, and the MFI for each photo was calculated. Statistical analysis was performed using a Student’s t-test.
Real-time RT-PCRTotal RNA was extracted from tissue samples and cells using TRIzol reagent (AG21102, Accurate Biology) after washing with PBS. According to the manufacturer’s instructions, cDNA was synthesized from purified RNA using an Evo M-MLV RT Mix Kit with gDNA Clean for qPCR (AG11728, Accurate Biology). SYBR Green PCR Master Mix (AG11718, Accurate Biology) was used for PCR amplification and a real-time PCR machine (ABI-7500, ThermoFisher) was used to quantify the expression of mRNAs. β-actin was used as an endogenous control, and the expression levels were quantified using the 2−ΔΔCt method. All primer sequences are listed in Additional file 2: Table S9, and each primer was detected in triplicate.
Western blottingCells and tissues were lysed by RIPA buffer (P0013C, Beyotime Biotechnology) with protease and phosphatase inhibitor cocktail (P002, NCM Biotech), and total protein concentration was measured using a bicinchoninic acid protein assay kit (YSD-500 T, Yoche). Samples were denatured by boiling in 1 × loading buffer and run on a 10% SDS–PAGE gel. Membranes were incubated with primary antibodies overnight at 4 °C, washed three times with TBST, incubated with secondary antibodies for 1 h at room temperature, and developed using Ultra High Sensitive ECL Kit (G2020-25ML, Servicebio). The primary and secondary antibodies used and their dilution are listed in Additional file 2: Table S8. Western blots shown in the accompanying figures are derived from three independent experiments.
Mouse studiesMouse Studies Athymic nude mice (BALB/cA nu/nu) aged 4 to 5 weeks (Shanghai Jiao Tong University School of Medicine, Shanghai, China) were housed in sterile cages under laminar airflow hoods in a specific pathogen-free room at 22–25° with a 12-h light and 12-h dark schedule and fed with autoclaved chow and water ad libitum.
In vivo subcutaneous xenograftFemale BALB/c nu/nu mice were purchased at 4 weeks of age and AtT20 cells were harvested and washed in PBS, resuspended with Hank's balanced salt solution (HBSS) about 1 × 106 (per side/100 μL) concentrations were injected subcutaneously on the suitable sites per mouse. Treatment was started when the tumor sizes reached approximately 100 mm3; mice were randomized into different groups. An intraperitoneal injection of Activin A (5 mg/kg in 100 μl PBS) or PBS was administered every other day. Tumor volumes (per group) were measured with a digital caliper and calculated as length × width2 × 0.52. Relative tumor volume (RTV) was calculated by (RTV = TVt / TV0, where TV0 is the tumor volume measured when starting drug treatment). The anti-tumor effect of drug treatment was calculated by drug-treated/control (T/C) ratio (T/C = RTVtreated / RTVcontrol × 100%). The mouse viscera were analyzed by HE staining to evaluate the safety of the inhibitor. The growth (Ki-67) and hormone (Pomc) of each tumor were measured by immunofluorescence (IF). The HE staining and IF analyses were performed, and the individuals performing the experiments were unaware of the samples being analyzed.
Statistical analysisAll statistical analyses were performed using R 4.2.1. All heatmaps were generated by the R package pheatmap (https://CRAN.R-project.org/package=pheatmap). The P-values were two-sided. P-values of less than 0.05 were considered statistically significant. For adjustment of P-values, P-values were adjusted using the Benjamini and Hochberg method (alias false discovery rate, FDR).
留言 (0)