
記住我
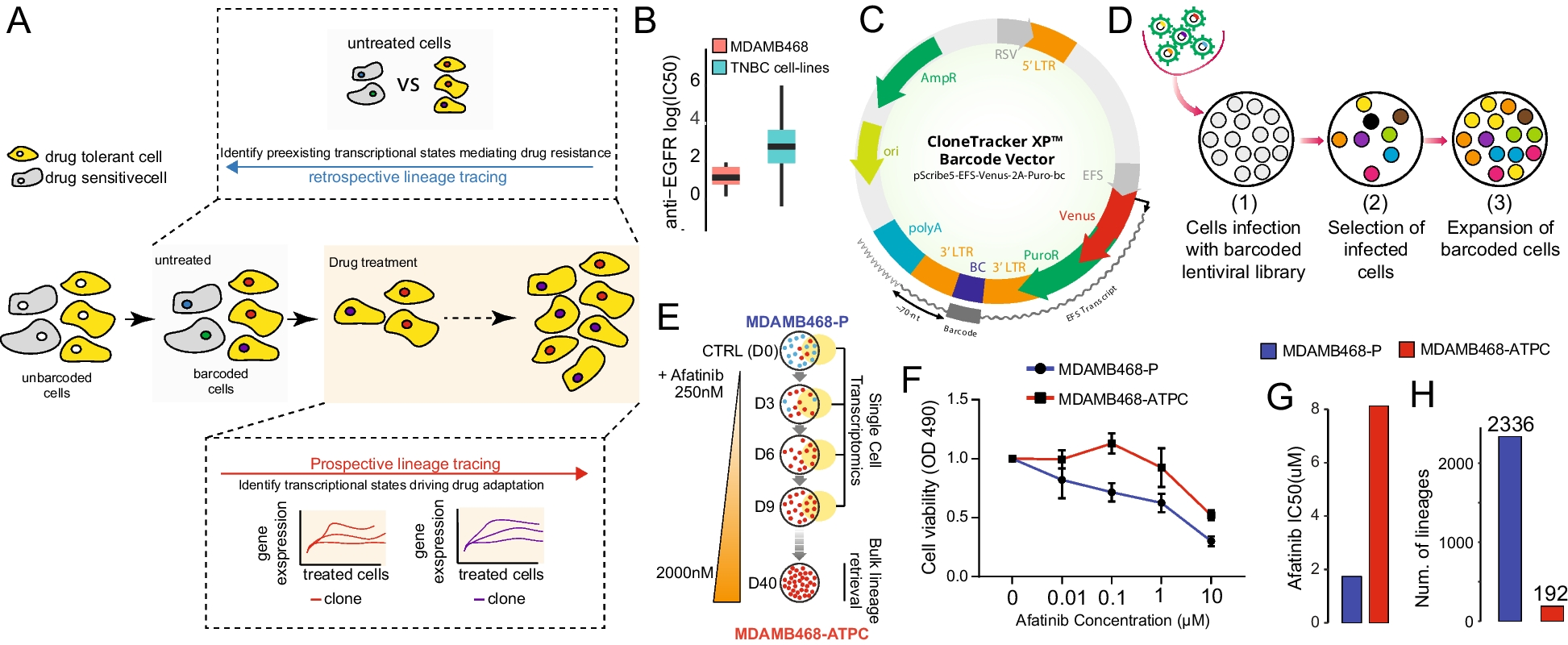
We developed a new likelihood-based method named dMTLV for detecting mtDNA variants of low VAF from WGS data. Briefly, dMTLV first performs a likelihood test to remove sequencing errors, then constructs consensus sequences via local assembly, and conducts another round of likelihood tests to remove PCR errors, finally generating highly confident mtDNA variants (Fig. 1a, Method).
Fig. 1
The workflow of dMTLV and the performance evaluation based on simulation data. a The workflow of dMTLV. For read family containing candidate alleles, dMTLV first performs a likelihood test to remove alleles likely to be sequencing errors, then constructs consensus sequences containing both true variants and PCR errors. Based on this, dMTLV performs a second likelihood test to eliminate PCR errors and generates the final result. b The TPR, PPV, and F1 of four tools: MToolBox, Mutect2, VarScan2, and dMTLV in detecting 20 randomly simulated SNVs under seven distinct VAFs and three different coverages. The comparison of VAF from 0.001 to 0.01 was highlighted with rectangles in light pink. c The Pearson correlation between the simulated and the observed VAFs by each tool across three different coverages. All the correlations were significant, with a p-value less than 0.01
We assessed the performance of dMTLV using 30 randomly simulated mtDNA variants under seven VAFs of 0.001, 0.005, 0.01, 0.05, 0.1, 0.25, and 0.5 and three sequencing depths of 5000 × , 10,000 × , and 20,000 × . Three existing tools: MToolBox, Mutect2 (“mitochondrial mode”), and VarScan2 were used for comparison using the quantifying indicators of TPR, PPV, and their harmonic mean F1 score. The correlation between the observed and the simulated VAFs of each variant for each tool was also compared (Methods).
Specifically, dMTLV achieved the highest TPR, PPV, and F1 for VAF < 0.01 across all three coverages in simulation data, but the PPV started to decrease from VAF of 0.1, suggesting that dMTLV is appropriate for detecting ultra-low VAF below 0.01. MToolBox achieved an intermediate TRP, PPV, and F1 across all VAF when the average coverages were 5000 × and 10,000 × but had the lowest PPV and thus the lowest F1 when the coverage reached 20,000 × , which is consistent with the initial report in the paper proposing MToolBox that it is appropriate for data with relatively low coverage. VarScan2 had the lowest TPR, PPV, and F1 when the VAF was 0.001, but performed intermediately when the VAF was larger across all coverages. Similarly, Mutect2 performs poorly when the VAF is below 0.01, but achieves the best result when VAF > 0.01 for all coverages, which is in line with its original design to detect mutations with a VAF greater than 0.01 (Fig. 1b).
For short insertion and deletions, across all coverages, Mutect2 had generally the lowest TPR for VAF < 0.01 but achieved a consistently high PPV for VAF > 0.01 for all coverages. By contrast, VarScan2 had the lowest PPV for both insertions and deletions across all coverages and VAFs. VarScan2 performed poorly for indels, especially for insertions. dMTLV performed the second best after Mutect2 for both insertions and deletions for all coverages and VAFs (Additional file 1: Fig. S1).
Mutect2 achieved the highest Pearson correlation for the VAFs between the simulated and the observed variants, followed by dMTLV and VarScan2. MToolBox had the lowest correlation probably due to its discarding anomalous read pairs when generating pileup files (Fig. 1c).
Collectively, dMTLV improved the TRP without sacrificing PPV for detecting mtDNA variants with VAF < 0.01, and the improvement gets more significant as the sequencing coverage increases. This result suggested that the integration of mtDNA variants detected by Mutect2 with VAF > 0.01 and those detected by dMTLV with VAF from 0.001 to 0.01 could capture the most accurate mtDNA variants covering the most comprehensive VAF ranges.
A new framework for non-ref NUMTs and NUMT-FPs detectionWe developed fNUMT for detecting non-ref NUMTs and the derived non-ref NUMT-FPs (Fig. 2a). Briefly, fNUMT takes the ref-NUMTs free bam file as input, of which the reads repetitively mapped to both nDNA and mtDNA were removed and extracts the soft-clipped reads with one portion of sequence mapped to nDNA and the remaining to mtDNA. These potential junction reads were then clustered to locate the insertion site of nDNA and the start and end positions of the inserted mtDNA segments. The latter is essential as the mtDNA are circular molecular with artificially defined coordination, unlike the segments in nDNA, of which the numerically smaller coordinate was naturally taken as the start position, the segments spanning the artificial break in the circular genome had a start position numerically larger than the end position. Nevertheless, the start and end positions were ambiguous in most previous studies, which confused the determination of segment size and the further exploration of the consequences of the non-ref NUTMs.
Fig. 2
The workflow of fNUMT and the performance evaluation. a (Left) The preprocess of WGS data for downstream analysis. The WGS clean data were first aligned to rCRS. The mtDNA-mapped reads were then aligned to the reference containing nDNA and rCRS to remove the candidate ref-NUMTs covered by reads that were repetitively mapped to autosome and rCRS. The ref-NUMT free data were subsequently input to fNUMT for detecting non-ref NUTMs and NUMT-FPs. (Right) fNUMT searches the candidate junction reads with one end mapped to nDNA and the other to mtDNA. Then cluster such soft-clipped reads according to the mapping coordination and direction to locate the breakpoints of the inserted mtDNA segments and the insertion site on nDNA. Next, fNUMT assembly all the reads mapped to the candidate mtDNA segments and realign the contig to mtDNA to confirm the occurrence of the non-ref NUTMs and detect the mismatches that were the potential non-ref NUMT-FPs. b The number of non-ref NUMTs detected by NUMTs-detection, dinumt, and fNUMT in the four samples. c, d An illustration of the confirmation of the non-ref NUMTs by long-read sequencing data, taking the non-ref NUMT chrM:59–16089-chr11:49883569 as an example. c The IGV plot of short-read data near chr11:49883569 where the soft-clipped reads that could also map to mtDNA (colored in blue) were observed, meaning the presence of insertion sequences originated from mtDNA near chr11:49883569. d The IGV plot of long-read data near chr11:49883569 where the insertion of sequences with inferred sizes of ~ 530 bp (red box) were observed. e The distance of the breakpoints estimated by NUMTs detection, dinumt an fNUMT over those inferred by long-read data. The p-value was measured by the Wilcox test
Next, fNUMT extracted and assembled the mtDNA-mapped sequences either from the potential junction reads or the discordant read pairs with one read mapped to mtDNA and the other to nDNA. The consensus sequence was then mapped to rCRS and the mismatched were detected as the potential non-ref NUMT-FPs.
To evaluate the performance of fNUMT, we applied it to the short read (150 bp, pair-end) WGS data of two ESCC paired tumor-normal samples and verified the results using the Nanopore long-read WGS data of the same samples [47]. The performance of fNUMT in detecting non-ref NUMTs was compared to the published methods NUMTs-detection [38] and dinumt [39]. The mean mtDNA coverage of these four samples was ~ 11,054, which is approximately five times higher than that of ~ 2000 × in the NUMTs detection paper. Therefore, when applied NUMTs-detection in our data, proportionally, we set the minimum supported discordant reads to 25 from the 5 used in the NUMTs-detection paper [38].
Specifically, the number of non-ref NUMTs detected by fNUMT, NUMTs-detection, and dinumt differed in all four samples (Fig. 2b). fNUMT identified one and four germline non-ref NUMTs for each patient, respectively. We verified all the ten non-ref NUMTs by the long-read sequencing data wherein we observed the insertion of sequences with inferred sizes near the estimated breakpoint of nDNA, and the inserted sequences could map to the coordinates of the inferred mtDNA segment (Fig. 2c, d, Additional file 1: Fig. S2, Additional file 2: Table S1). Of note, two non-ref NUMTs verified by long-read data were not detected by NUMTs-detection and dinumt, indicating that fNUMT had better specificity. Moreover, we ranked the non-ref NUMTs identified by NUMTs detection and dinumt according to the confidence decided by the number of supported signals and the quality score, separately, and observed that even the most confident one cannot be confirmed by the long-read sequencing data (Additional file 1: Fig. S3).
Taking the breakpoints of long-read data as the gold standard, we next evaluated the precision of breakpoint estimation of fNUMT, NUMTs-detection, and dinumt using the eight shared non-ref NUMTs. Each non-ref NUMT had three (or four if the estimated two nDNA positions were not the same) breakpoint distances calculated by the absolute differences between the breakpoint estimated by each tool and the long-read data. Overall, fNUMT predicted the most precise breakpoints. The mean breakpoint distances of fNUMT, dinumt, and NUMTs-detection were 2.31 bp, 88.63 bp, and 154.56 bp, respectively (Fig. 2e). Take the non-ref NUMTs: chrM:59–16089-chr11:49883569 in tes200124-T-N-1 as an example, of which the inferred mtDNA segments by fNUMT was chrM:61–16089, the same with that estimated by long-read data while the chrM:16090–16489 inferred by NUMTs-detection missed chrM:1–61 and chrM:16490–16569. In terms of the insertion site on nDNA, the chr11:49883569–49883572 estimated by fNUMT was 0 bp and 2 bp away from the long-read decided breakpoint of chr11:49883569 while the chr11:49883298–49883856 by NUMTs-detection were 271 bp and 287 bp away, and chr11:49883500–49883665 by dinumt were 69 bp and 97 bp away. More specifically, among the inferred 30 breakpoints of all 10 non-ref NUMTs detected by fNUMT, 11 were the same, 10 with the distance of 1 bp, 4 with 2 bp, 2 with 3 bp, and 2 with 6 bp, indicating that the breakpoints inferred by fNUMT were very close to that identified by long-read sequencing data (Additional file 2: Table S1).
In addition, fNUMT identified 22, 24, 28, and 27 potential non-ref NUMT-FPs in each of the four samples, among which two were indels present in all samples (m.16258_16259insA and m.16264_16264del) that accumulated in the non-ref NUMT chrM:61–16089-chr11:49883569–49883572. As non-ref NUMT-FPs were not generated by NUMTs-detection and dinumt, the performance regarding non-ref NUMT-FPs could not be compared.
Multi-omics data of ESCC and the mtCN characterizationIn this study, we included the WGS data of 663 ESCC paired tumor-normal samples, of which the RNA-seq and WGBS were also available in 155 patients [23, 24]. The mtCN was estimated by the sequencing depth of mtDNA (normal: 11,957.1 × , tumor: 11,108.2 ×) and nDNA (normal: 29.6 × , tumor: 62.5 ×), which showed a significant positive correlation in each sample type (Fig. 3a). We further adjusted the mtCN in tumor samples based on the purity and ploidy estimated by ABSOLUTE [50]. On average, the mtCN of tumor samples was 412.3, significantly lower than 808.5 in normal samples (p < 2e − 16). At the individual level, 592 out of 663 (89.3%) patients had decreased mtCN in the tumor compared to the paired samples, while the remaining 71 patients had the opposite trend (Fig. 3b).
Fig. 3
The multi-omics feature of mtCN in 663 ESCC paired tumor-normal samples. a The sequencing coverage of mtDNA and nDNA in the 1326 patients. The correlations were evaluated by the Pearson test. b The mtCN in normal and tumor samples. Lines colored in blue indicate a lower mtCN in the tumor over the paired normal samples, otherwise in purple. The p-value was calculated by the two-sided t-test. c The Spearman correlation between the expression of the mtDNA coding genes with mtCN in the tumor, normal, and all samples. d The Spearman correlation between the methylation level of the mtDNA regions with mtCN in the tumor, normal, and all samples. e Plots of overall survival for ESCC patients with mtCN above and below the median value. f Plots of overall survival for ESCC patients with TN-ratio above and below 1. For both e and f, the shaded areas represent the 95% confidence intervals, with the risk table under the survival plot. p values were calculated by log-rank test. g The TN-ratio and mtCN under different T stages. The pairwise comparison was performed by the Wilcox test (* < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001, ns not significant), and the overall p values were calculated by the one-way ANOVA test. h The differentially expressed genes of the tumor samples of patients with TN-ratio > 1 over those with TN-ratio < 1. p values were calculated by the Wilcox test and adjusted by the Bonferroni method. i, j The enriched GO terms of genes significantly (p < 0.01) upregulated (i) and downregulated (j) in the tumor samples of patients with TN-ratio > 1. The p values were adjusted by the Bonferroni method
We next evaluated the relevance of mtCN to gene expression and methylation. Specifically, we found a significant positive correlation between the expression of MT-CO1, MT-CO3, MT-ND3, and MT-RNR2 with mtCN in all samples, while a negative association was found in MT-ND6 (Fig. 3c). Regarding the methylation level of all mtDNA regions, we observed a significant positive correlation of the methylation level of MT-CO3, MT-RNR1, and five tRNAs (particularly tRNA-Thr) with mtCN in tumor samples; however, this was not found in normal and all samples (Fig. 3d).
Previous mtDNA studies in pan-cancer have shown that a high mtCN in tumor tissue is associated with a better prognosis in some cancer types and an inverse trend in others [14]. In this study, the patients with high mtCN in tumor samples tended to have worse overall survival; however, this was only marginally significant (p = 0.063, Fig. 3e).
Instead, we observed a significant association (p = 0.0015) between the overall survival with the TN-ratio calculated as the ratio of mtCN between the paired tumor and normal sample (Fig. 3f). Specifically, patients with TN-ratio > 1 had worse overall survival, suggesting that the tumor cells in these patients may have a better ability to compete for individual energy than normal cells, thus leading to a worse prognosis. Given the differences in energy metabolism basis between individuals, the comparison of TN-ratio would be more reasonable than simply comparing the mtCN of tumor tissues among patients.
Interestingly, we observed that the TN-ratio increased with the T stage progressed (p = 0.00057), which was mainly ascribed to the increase of mtCN in tumor samples (p = 0.005), while the mtCN was nearly unchanged in normal samples (Fig. 3g). This result was similar to the previous finding that mtCN increased as diseases progressed from noncancerous esophageal mucosa to ESCC and finally to lymph node metastasis [25]. Since patients with high T stage tend to have a worse prognosis, this result supported the correlation between TN-ratio and overall survival from another perspective. Except for the T stage, we did not observe significant associations between TNratio with other clinical features.
We further explored the genome-wide differentially expressed genes between patients with TN-ratio > 1 (group 1, patient number: 19) and those with TN-ratio < 1 (group 2, patient number: 136; Fig. 3h). We observed that the up-regulated genes in group 1 were enriched in energy metabolism-related pathways, suggesting that the tumor cells in these patients had better metabolic capacity (Fig. 3i). On the contrary, many of the down-regulated genes are histone modification-related genes. As some of the intermediates of mitochondria are the substrates of chromatin modification enzymes in the nucleus, the down-regulated expression of histone modification-related genes may be caused by the regulation of mitochondrial intermediates, which may increase chromatin accessibility and thus promote the proliferation of tumor cells. Other down-regulated genes were significantly enriched in cell cycle checkpoint and DNA damage repair-related pathways (Fig. 3j), suggesting that the tumor cells in group 1 were more capable of escaping the cell cycle monitoring and damage repair, which would be relevant with the malignant proliferation of tumor cells which thus lead to worse survival.
The profile of non-ref NUMTs in ESCCWe applied fNUMT to detect the non-ref NUMTs of 663 ESCC paired tumor-normal samples based on the short-read WGS data. At the sample level, the average number of non-ref NUMTs in normal samples was 2.1, slightly lower than 2.4 in tumor samples (Fig. 4a). Overall, 1227 (92.53%) had at least one non-ref NUMTs. More specifically, 1166 (87.93%) had at least one germline non-ref NUMTs; 249 (40.62%) tumor samples had at least one somatic non-ref NUMTs, and 98 (15.99%) patients had at least one non-ref NUMTs present in normal sample but absent in the paired tumor sample (loss) (Fig. 4b).
Fig. 4
The landscape of non-ref NUMTs in 663 ESCC patients. a The number of non-ref NUMTs per sample. The dashed lines indicate the average number of non-ref NUMTs per sample type. b The number of non-ref NUMTs of the germline, loss, and somatic per individual. c The correlation between the frequency and size of the 110 distinct non-ref NUMTs detected in all 1326 samples, with summary histograms at the edges. The high-frequent non-ref NUMTs may include more than one type of source indicated by different colors and shapes. The distribution of non-ref NUMTs with frequency < 0.2 and size < 2500 bp was zoomed in. d The circos plot of 110 distinct non-ref NUMTs. From the outside: (1) mtDNA genes (left) and nDNA chromosome (right); (2) frequencies of non-ref NUMTs reported in Wei W. et al. [38]; (3) frequencies of non-ref NUMTs reported in Dayama G. et al. [39]; (4) frequencies of non-ref NUMTs detected in this study; and (5) arrows representing the insertion of mtDNA segments to the breakpoints of nDNA. e The number of somatic simple (left) and complex (right) SVs in 528 tumor samples with (num = 468) and without (num = 60) non-ref NUMTs. p values were calculated by the Wilcox test. f The circos plot of 46 distinct somatic non-ref NUMTs. g The circos plot of 22 tumor specific non-ref NUMTs. The genes with non-ref NUMTs on the intron region were labeled. Only the frequency in our cohort was shown in f and g. h (Left, middle) The expression of CBWD1 in different sample types in patients with different sources of the non-ref NUMTs chrM:6225–6420-to-chr9-129767. (Right) The expression of CBWD1 in samples with and without the non-ref NUMTs. p values were calculated by the Wilcox test
At the population level, we identified a total of 110 distinct non-ref NUMTs, 85 (77.3%) of which were < 1 kb and present in < 10% of 663 ESCC patients (Fig. 4c). On average, the size of non-ref NUMTs in tumors (470.47 bp) was slightly larger than in normal samples (443.26 bp). Interestingly, we observed that the most frequent four non-ref NUMTs had small sizes (< 100 bp), and all were present as germline. On the contrary, the 14 non-ref NUMTs with the largest sizes had low frequencies (< 5%) and were mostly (13, 92.9%) somatic or loss. These results implied that the non-ref NUMTs may have potential roles in ESCC and thereby underwent different selection pressures.
We also compared the 110 non-ref NUMTs with those (num = 1601, Methods) identified in 66,083 human genomes from 100,000 Genomes Project in England [38], and in 946 and 53 genomes from 1 KGP and HGDP, respectively [39] (Fig. 4d). Generally, the insertion sites of the 110 distinct non-ref NUMTs were distributed across the whole genome; the most frequent non-ref NUMTs (> 10%) occurred on chr11, chr9, chr4, chr3, chr2, and chr1, with corresponding mtDNA region of D-loop, MT-COX1, MT-ND5, and MT-CYTB (shared by the last three), which was similar with previous finding. Most (94, 85.5%) of the non-ref NUMTs were < 1 kb (Additional file 1: Fig. S4). Overall, 24 non-ref NUMTs were also identified previously. When ranking the 110 non-ref NUMTs according to the population frequency, all the top 13 non-ref NUMTs (> 10%) were also found in previous studies with comparable population frequencies (Additional file 2: Table S2). The most frequent non-ref NUMT, present in 69.83% of samples, was the insertion of the 541 bp D-loop segment (chrM:16089–61) spanning the artificial break to chr11:49883569, which would otherwise be mistaken as chrMT:61–16089 with the size of 16,027 if the start and end positions were not accurately determined.
Noteworthy that one non-ref NUMT (chrM:12714–14830-to-chr5:32338477 spanning MT-ND5, MT-ND6, and MT-CYTB) identified previously with the East-Asian frequency of 68% and the 1 KGP and HGDP frequency of 25.93% was absent in our cohort (Fig. 4d). This non-ref NUMT was also detected by NUMTs-detection and dinumt in the two ESCC paired samples used for fNUMT performance evaluation. However, as indicated by the long-read sequencing data, the size of the sequence inserted to chr5:32338477 was 293 bp rather than 2116 bp (Additional file 1: Fig.S5). Furthermore, this 293 bp sequence was the “Homo sapiens chr5:32338477–32338478 non-reference unique insertion sequence” (NCBI accession number: MH534373), of which the 1–147 bp aligned to chrM:14832–14987, and the 147–190 bp reversely aligned to chrMT:12723–12867, leading to the wrong identification of the insertion of chrM:12714–14830. By contrast, fNUMT managed to avoid such false positives by clustering potential junction reads according to their mapping orientation and filtering out candidate non-ref NUMTs with orientation conflict or only one type of orientation cluster.
Potential consequences of non-ref NUMTs in ESCCWe next investigated the association of non-ref NUMTs with the previously dissected somatic structural variants (SVs) in the nDNA of 528 ESCC individuals (out of the 663 in this study) [47]. Interestingly, we observed that the tumor samples carrying at least one non-ref NUMTs (num = 468) tend to have more SVs that were defined as complex than those without any non-ref NUMTs (num = 60, p = 0.013, Fig. 4e), implying that the insertion of mtDNA sequences to nDNA may affect the stability of nDNA thus leading to complex SVs. As expected, this trend was not found for simple SVs, meaning that the initiation of simple SVs was not affected by non-ref NUMTs (Fig. 4e).
Regarding the origins of non-ref NUMTs, we identified 46 distinct somatic non-ref NUMTs with a mean size of 952.45 bp (Fig. 4f). The most frequent somatic non-ref NUMTs (present in 85 patients) was the insertion of 195 bp segments in COX1 to the intronic region of CBWD1 (chrM:6225–6420-to-chr9-129767). Among the somatic non-ref NUMTs, 22 (present in 23 patients) were ESCC tumor-specific absent in any normal samples of our cohort and the previous study [38]. Five such non-ref NUMTs were in the intron region of FAF1, PDS5B, RECQL5, KIF16B, and GABRA3, and 1 in the UTR3 of MX1 (Fig. 4g).
Among the 75 germline non-ref NUMTs (Additional file 1: Fig. S5), the one on the intronic region of TCF12 (chrM:7179–7295-to-chr15:57220850, present in one individual) would be interesting as TCF12 is a known tumor-associated gene (COSMIC databases, [53]). Additionally, as the insertion of mtDNA to the exon region is very rare, one germline non-ref NUMT (chrM:11707–11854-to-chr8-139661937, present in one individual) on the exon region of COL22A1 would also be appealing.
We next explored the potential effect of non-ref NUMTs on the expression of the host or nearest genes. We evaluated the 11 non-ref NUMTs present in at least 5 samples of 155 patients with available RNA-seq data. The result showed that the non-ref NUMT chrM:6225–6420-to-chr9-129767 on the intron region of CBWD1, present in 153 out of 310 samples (21 somatic, 126 germlines, and 6 loss), downregulated the expression of CBWD1 particularly in normal samples (Fig. 4h). Regardless of whether this non-ref NUMT exists, the expression of CBWD1 was significantly lower in tumors (34.24) than in normal samples (42.09). However, this non-ref NUMT further reduced the expression of CBWD1 in normal samples from 46.36 to 36.75, with the latter comparable to that of tumor samples. Moreover, the expression of CBWD1 was further reduced in tumor samples with the non-ref NUMTs. In fact, CBWD1 enables ATP binding activity and is active in the cytoplasm [57], which may be the reason of the high frequency of this non-ref NUMT. The downregulation of CBWD1 may affect the ATP binding activity and the subsequent ATP production, which may modulate the cellular energy metabolism, and thereby providing conditions for tumor cells to adapt to a hypoxic environment.
The landscape of mtDNA variants in ESCCWe applied dMTLV and Mutect2 to detect mtDNA variants of the 663 ESCC paired tumor-normal samples from the ref-NUMT free bam files described in Fig. 2a. We obtained an average of 1070 initial mtDNA variants per sample by integrating mtDNA variants with VAF > 0.01 detected by Mutect2 and those with VAF from 0.001 to 0.01 detected by dMTLV. In addition, we identified an average of 76 potential non-ref NUMT-FPs (Additional file 1: Fig. S6; Method) for each sample. After removing non-ref NUMT-FPs, we observed a strong negative correlation between the number of mtDNA variants with VAFs ranging from 0.001 to 0.01 and mtDNA coverage (R = − 0.34, p < 2.2e − 16, Additional file 1: Fig. S7), suggesting the existence of depth-related false positive variants among these low-frequent mtDNA variants. As the variants should theoretically appear in at least one mitochondrion, those with VAF below 1/mtCN were supposed to be false positives. After removing these variants, the number of variants with VAF from 0.001 to 0.01 was in proportion with mtDNA coverage (R = 0.26, p < 2.2e − 16, Additional file 1: Fig. S7), consistent with the rationale that the increase of sequencing coverage improves the chance of identifying low-frequent altered alleles. As expected, the detection of mtDNA variants with VAF above 0.01 was rarely affected by mtDNA coverage, indicated by the similar R score before and after mtCN-based filtering (Additional file 1: Fig. S7).
The above pipeline of mtDNA variants detection and filtering resulted in an average of 214.5 variants in tumors, significantly lower than 521.9 in normal samples (p < 2e − 16, Fig. 5a), consistent with the hypothesis that tumor cells were able to eliminate defective mitochondria (with mtDNA variants) to keep energy homeostasis to survive the changing micro-environment during tumor initiation and progression [58]. Additionally, the number of mtDNA variants was in proportion with mtDNA coverage, suggesting the robustness of the mtDNA variants.
Fig. 5
The profiling of mtDNA variants in ESCC. a The correlation between the number of mtDNA variants and coverage in normal and tumor samples, with the summary boxplot in the margin. The relation coefficients and p values were calculated by the Pearson test. b The circos plot of the population frequency of the 24,347 distinct mtDNA variants detected in the 1326 samples. From the outside: (1) the position of the mtDNA; (2) the regions of mtDNA; (3) the frequency of all, germline, loss, and somatic mtDNA variants; and (4) the frequency of somatic mtDNA variants in the MITOMAP database. c The correlation between the max VAF of truncating mutations with mtCN in tumor and normal samples, with the summary boxplot showing in the margin. The relation coefficients and p values were calculated by the Pearson test. d Plots of overall survival for patients with mean VAF of truncating mutations of tumor sample above and below the median value. The colored areas indicate the 95% confidence intervals, with the risk table under the survival plot. p value was measured by log-rank test. e The proportion of six mutational types under different VAFs, with the mean number per type shown on the right. f Five mutation signatures extracted from the somatic mtDNA variants. g The cosine similarity between the five signatures in our cohort with COSMIC single base substitution signatures. h The correlation between the number of truncating mutations with the contribution of the five mutation signatures. The relation coefficients and p values were calculated by the Pearson test
At the population level, we identified 24,347 distinct mtDNA variants. The hotspot regions include D-loop, MT-ND1, MT-ND4, and MT-ND5 regardless of the source of the variants (germline, loss, or somatic, Fig. 5b). Among these, 10,316 (42.4%) variants were also reported in MITOMAP database (the overall population cohort), and 700 (2.88%) were previously identified as disease-associated variants, of which 73 were experimentally confirmed. Interestingly, 640 (2.63%) were reported as somatic variants of many cancer types wherein three variants in D-loop (215A > G, 16304 T > C, 16324 T > C) have been observed in EC. The somatic variants identified in our cohort and those found in cancers of MITOMAP both showed enrichment on D-loop and MT-ND4, while our results were additionally enriched on MT-ND1 and MT-ND5 (Fig. 5b). Additionally, we identified 126 ESCC tumor-specific variants (62 SNVs, 49 short insertions, and 15 short deletions) present in at least two tumors but not in any normal samples nor MITOMAP general populations (Additional file 2: Table S3).
We next evaluated the potential contribution of truncating mutations (stop-gain and frameshift indels) to ESCC. We observed that the per-sample max VAF of truncating mutations (t-VAF) in tumors was significantly higher than in normal samples (mean: 0.044 versus 0.0068, p = 2.25e − 19). Moreover, samples with max t-VAF above 0.11 were all tumors (num = 55), implying that tumor cells were more capable of accumulating mtDNA variants that were functionally advantageous while eliminating the damaged mtDNA, with the latter indicated by the reduced mtCN in tumors (Fig. 5c). More interestingly, we observed that patients with higher mean t-VAF in the tumor samples had better overall survival (p = 0.008, Fig. 5d), suggesting that tumors with more comprehensive elimination of dysfunctional mtDNA tended to have better survival.
The primary mutation types of mtDNA SNVs in our cohort were T > G, T > C, and C > T, with the mean proportion of 32.14%, 27.83%, and 23.89%, respectively (Fig. 5e, Additional file 1: Fig. S8, Additional file 1: Fig. S9a), which was different with other cancer types reported in previous studies wherein the C > T and T > C had the first and second largest proportion and T > G account only for a small proportion [14]. As this previous study only reported mtDNA variants with VAF > 0.01, we further analyzed the mutation types under different VAFs and observed that the mutation types of variants with VAF > 0.01 were also dominant by C > T and T > C, and the percentage continued to increase for variants with VAF > 0.05 and 0.1 (Fig. 5e). Interestingly, we observed worse overall survival for patients with a higher number of low-frequent T > G variants in tumor samples (p = 0.01, Additional file 1: Fig. S9b), indicating that these large percent T > G variants in our cohort may reflect the instability of mtDNA and thereby affect the prognosis of ESCC.
We next investigated the mutational signature of all somatic variants, which could be classified into five distinct signatures by the NMF method (Fig. 5f). The cosine similarity against COSMIC single base substitution (SBS) signatures (v3.3-June 2022) showed that Signature_MT3 significantly correlated with SBS28 (unknown) with a cosine similarity of 0.857, Signature_MT4 associated with SBS32 (Azathioprine treatment), SBS30 (Defective DNA base excision repair due to NTHL1 mutations), and SBS11(Temozolomide treatment), with similarity of 0.887, 0.839, and 0.805, respectively; Signature_MT2 marginally correlated with SBS22 (aristolochic acid exposure) with similarity of 0.594 and Signature_MT5 marginally associated with SBS37 (unknown) with similarity of 0.635. Signature_MT1 was supposed to be novel without obvious correlation with any COSMIC SBS signatures (Fig. 5g). Interestingly, we found that the number of truncating mutations was in proportion with the contribution of Signature_MT2 and Signature_MT4 (Fig. 5h), reflecting the potential association of mtDNA truncating mutation with aristolochic acid exposure, DNA base excision repair, azathioprine, and temozolomide treatment, all of which were relevant to tumorigenesis and treatment response.
留言 (0)