
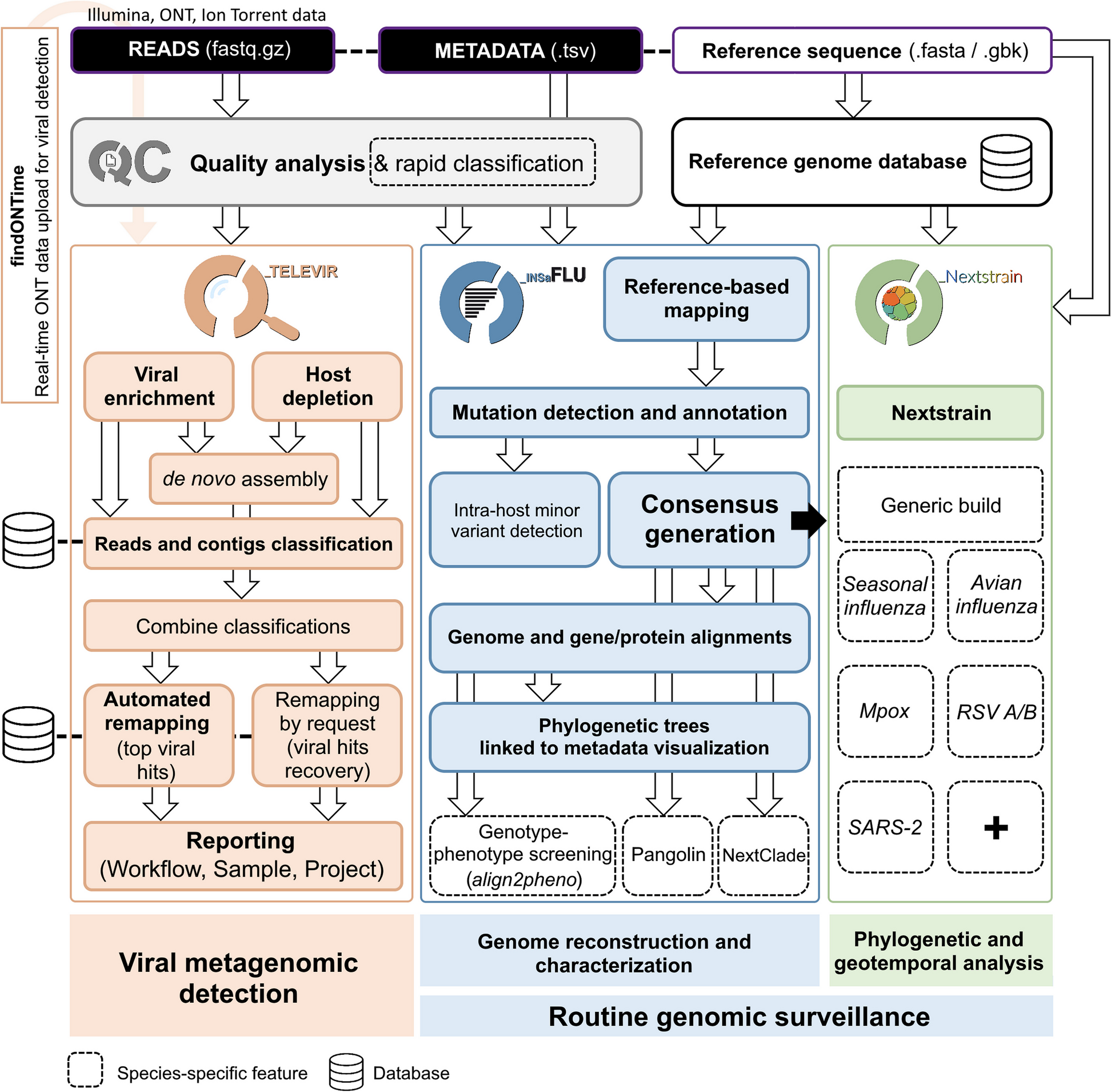
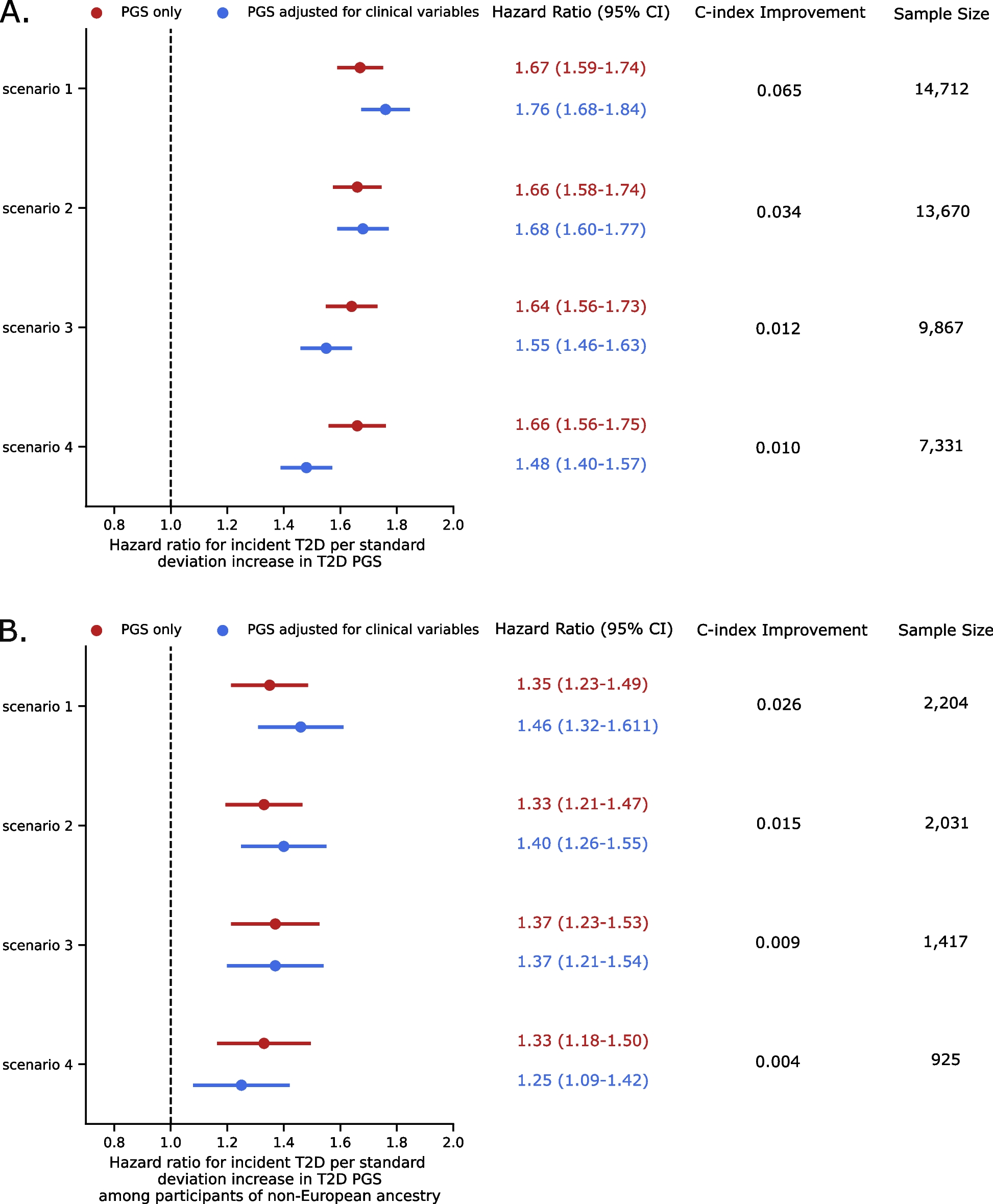
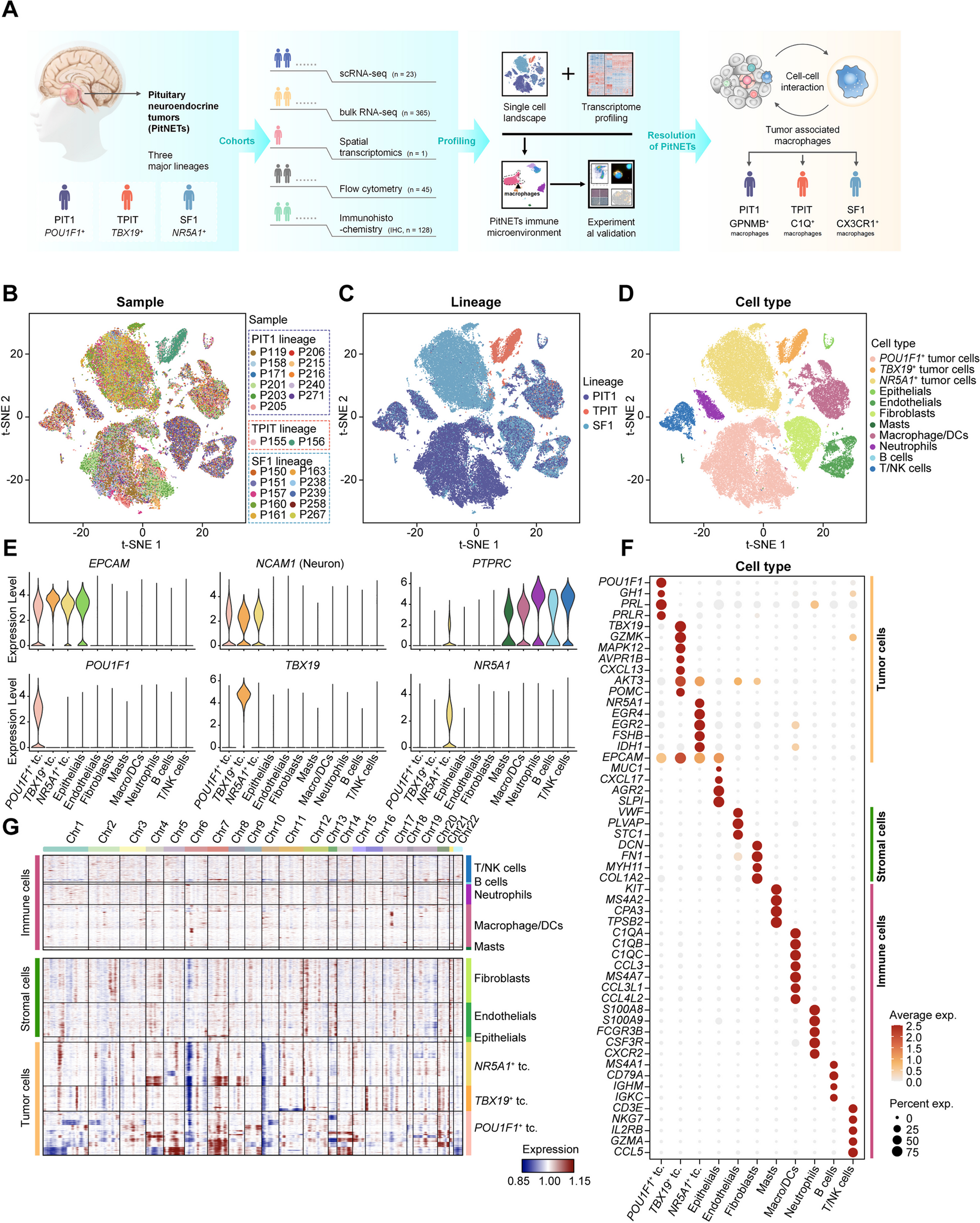
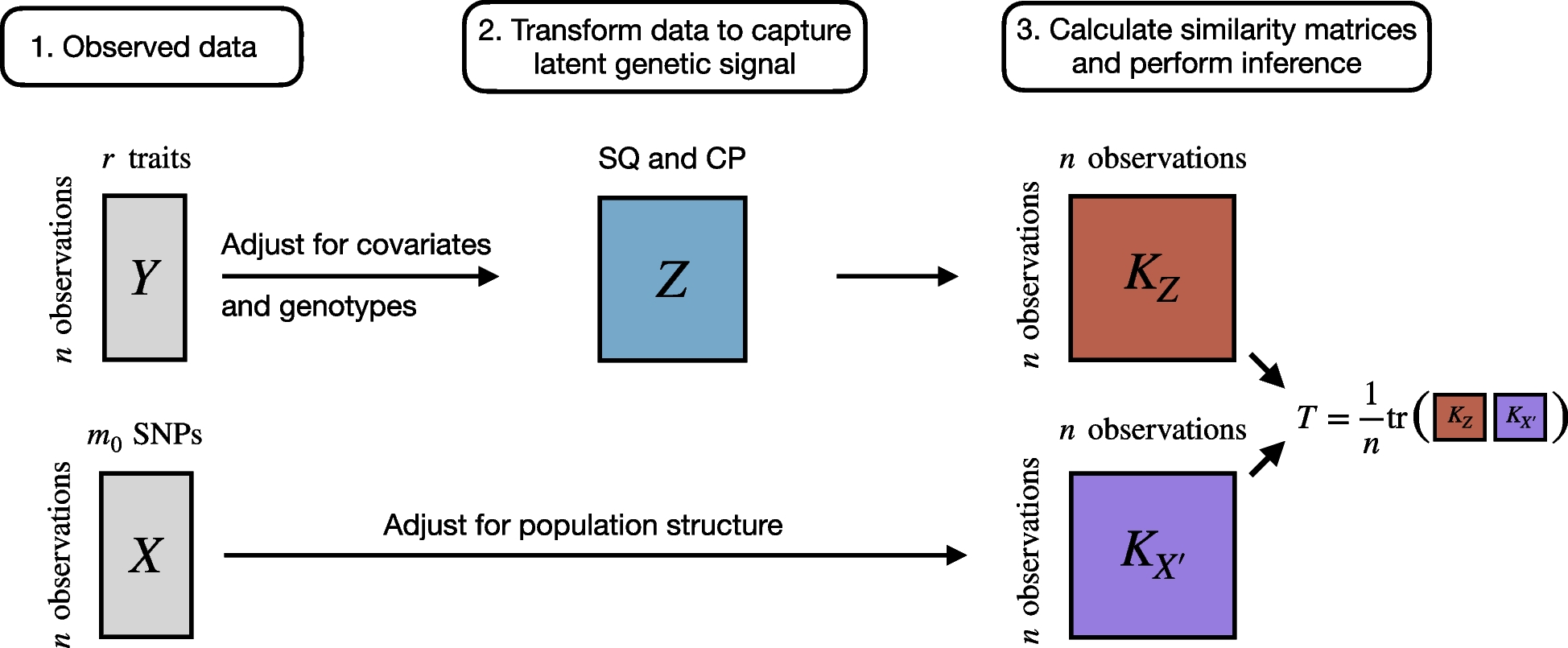
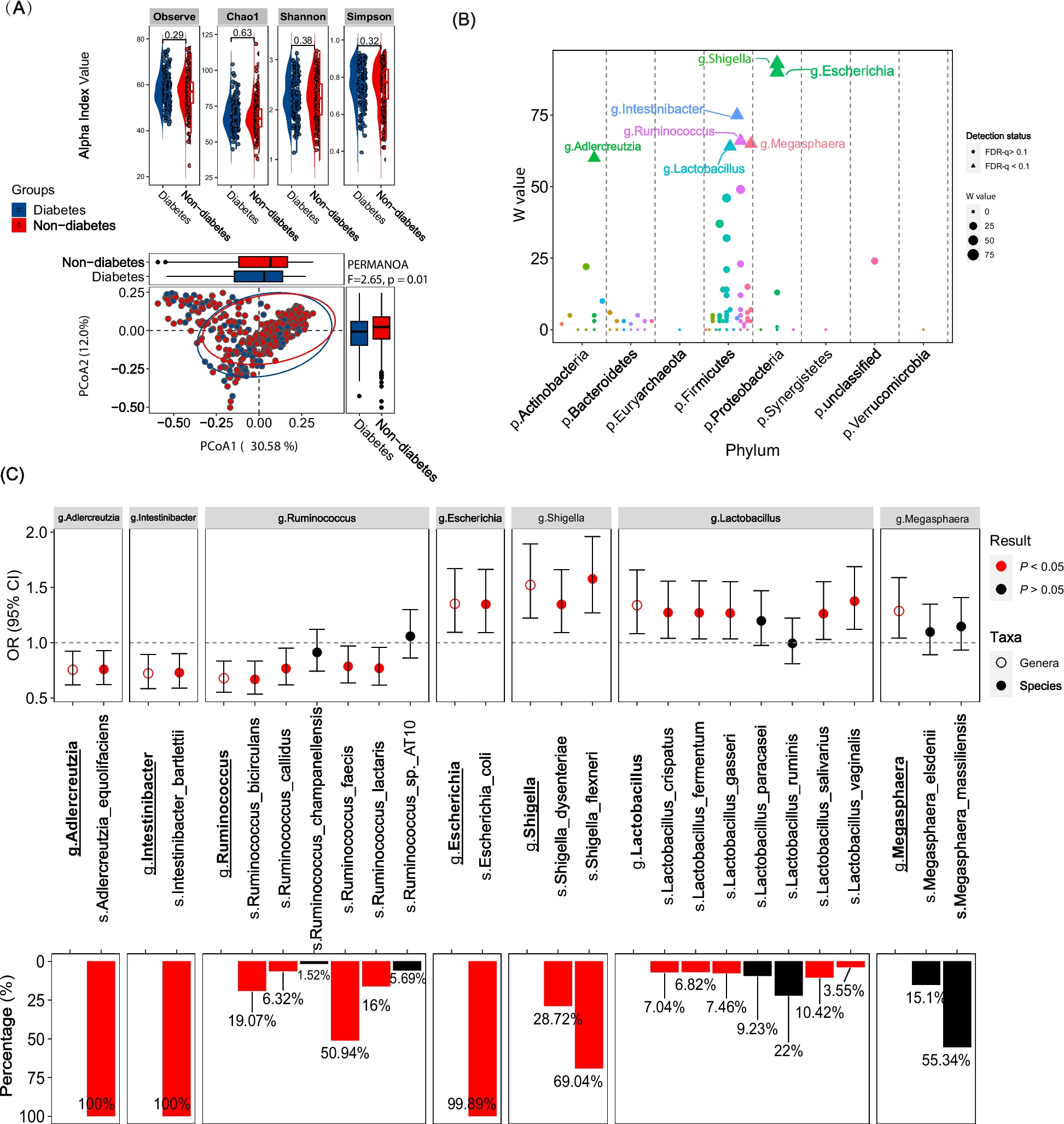
記住我
We constructed a workflow to derive conserved DNA methylation signatures (Fig. 1A). We first analyzed 569 paired methylation profiles of both tumor and matched normal tissues from The Cancer Genome Atlas (TCGA) (Additional file 1: Supplementary Methods). We retrieved 253,574 differentially methylated positions (DMPs) based on the significance of differences in beta values (∆\(\beta\)) between matched tumor and normal tissue in nine cancer types (permutation test, P < 0.001) (Methods, Additional file 1: Fig. S1, Additional file 2: Table S1). Among the significant DMPs, 129,868 were hypermethylated, and 122,782 were hypomethylated in cancer patients. We further selected the most conserved significant Hyper-DMPs (N = 2969) and Hypo-DMPs (N = 4395) by controlling the variation in ∆\(\beta\) (Additional file 1: Supplementary Methods; Additional file 1: Fig. S1). To verify the DMPs, we compared the methylation levels of the top 10 Hyper and Hypo-DMPs between cancer and normal tissues in 9 cancer types and showed that the DMPs were indeed significantly differentially methylated among the cancer types (Additional file 1: Fig. S2 and S3). We characterized the genomic locations of Hyper and Hypo-DMPs and found that most of the Hypo-DMPs (71.07%) were located in the Open Sea region, while the majority of the Hyper-DMPs (67.03%) were located at CpG islands (Additional file 1: Fig. S4A-B), suggesting a general tendency toward reprogramming of the cancer methylome. We then examined the binding activities of known regulators of DNA methylation, namely, DNMT1, DNMT3B, and TET2, at the DMPs [32, 40]. Of the three genes, TET2-binding activity was selectively enriched at Hypo-DMPs in MCF-7 cells (Additional file 1: Fig. S4C), while DNMT1 and DNMT3B-binding activity were not enriched at the DMPs (Additional file 1: Fig. S4D, E).
Fig. 1
Workflow of deriving DNA methylation signatures in cancers
To further understand the variations in the conserved DMPs in cancer, we applied nonnegative matrix factorization (NMF) to the methylation profiles of Hyper-DMPs (n = 2,969) and Hypo-DMPs (n = 4,395) in 2,465 TCGA cancer samples representing nine cancer types. As a result, we identified three distinct hypermethylation signatures (hereafter, Hyper-MS1-3) and seven hypomethylation signatures (hereafter, Hypo-MS1-7) (Methods) based on cophenetic coefficients and silhouette coefficients (Additional file 1: Fig. S5A). Each methylation signature was defined as a positive linear combination of a distinct set (321 to 1231) of contributing DMPs. The contributions of each DMP to a given methylation signature (probe weight, E) were all nonnegative and summed to one (Additional file 1: Fig. S5B). For comparison, we also derived hypomethylation signatures using 1-β values, namely, Hypo-MSs-down, and found that the resulting signature scores were nearly identical to the original Hypo-MSs derived from β values (Methods, Additional file 1: Fig. S6).
The landscape and characteristics of the DNA methylation signaturesWe found that the activities of DNA methylation signatures are highly variable in the cancer population (Fig. 2A and Additional file 1: Fig. S7A). The majority (78.09% ~ 99.63%) of the variation was attributed to the different tissue origins of cancer (Additional file 2: Table S2; Additional file 1: Supplementary Methods). Our data showed that all cancer types exhibited one to eight predominant methylation signatures. Some methylation signatures are cancer type specific. For example, Hyper-MS3 and Hypo-MS2 are active specifically in COAD, and Hypo-MS3 activity is specific to PRAD. In contrast, several methylation signatures, such as Hypo-MS4 (HNSC, LUAD, LIHC, BRCA, KIRP, and KIRC) and Hyper-MS1 (BRCA, HNSC, LIHC, PRAD, KIRC, KIRP, and THCA) (Fig. 2A), were active in multiple cancer types. In addition, the DMPs contributing to the hyper and hypomethylation signatures exhibited distinct patterns of localization (Additional file 1: Fig. S7B), suggesting a general tendency toward reprogramming of the cancer methylome.
Fig. 2
Characterization of DNA methylation signatures in cancers. A Proportion of each cancer type in which different DNA methylation signatures are present. B Cosine similarity between different methylation signatures (MSs). C Cosine Similarity and Spearman correlation between the probe weight of methylation signatures derived from cancer cell lines (CC-MSs) and tumor tissues (TCGA-MSs). The top 20 weighted probes in each signature are labeled with the corresponding gene names.
The activities of the Hyper-MSs (cosine similarity, CS ≤ 0.31) and the Hypo-MSs (CS ≤ 0.24) (Fig. 2B and Additional file 1: Fig. S7C) in the TCGA population were highly independent, suggesting that these signatures represent distinct biological activities. To further verify the conservativity and consistency of the methylation signatures derived from heterogeneous tumor tissues, we performed NMF on the methylation profiles of the same set of DMPs used to derive the hyper and hypomethylation signatures in 241 cancer cell lines representing the same nine cancer types (Additional file 1: Supplementary Methods). We obtained four cancer cell-based hyper and hypomethylation signatures (CC-Hyper-MS1 and 2, CC-Hypo-MS1 and 2) (Additional file 1, Fig. S7D). We evaluated the consistency of the probe-weight matrix E between cell-based methylation signatures and tissue-based signatures (Additional file 1: Supplementary Methods). The TCGA-derived Hypo-MS1 (tumor tissue) was highly consistent with CC-Hypo-MS1 (CS = 0.79, Spearman’s R = 0.45) and TCGA-Hypo-MS4 consistent with CC-Hypo-MS2 (CS = 0.88, Spearman’s R = 0.63). In addition, TCGA-Hyper-MS2 resembled CC-Hyper-MS1 (CS = 0.83, Spearman’s R = 0.59) (Fig. 2C). Among the most conserved and highly weighted genes between the cell-based and tumor methylation signatures were many known oncogenes, such as ETS1, RORA, and CDKN1A (Hypo-MS1); immune genes, such as SIGLEC9, SIGLEC7, MUC15, and LAIR1 (Hypo-MS4); and SEPT9 [49] and DDIT3 [50] (methylation-related biomarkers) for Hyper-MS2. These findings suggest that Hyper-MS2, Hypo-MS1 and Hypo-MS4 are driven by conserved biological processes in cancers.
DNA methylation signatures are associated with clinical featuresMany previous studious have shown the methylome correlates with many clinical and pathological features [51, 52]. Therefore, we verified the relationship between our methylation signatures and clinical outcomes. We divided the patients into two groups (low and high) according to the median methylation signature activity and then investigated the associations among the groups and patient age, overall survival, and cancer stage within individual cancer types (Methods). As a result, most of the Hypo-MSs (6/7) showed no association with patient age in the cancer types where they were present, except for Hypo-MS5, which was negatively associated with age in the THCA (FDR = 0.00738) (Wilcoxon rank-sum test). For the Hyper-MSs, Hyper-MS1 and Hyper-MS2 exhibited significant correlations with age in BRCA and THCA (Hyper-MS1: FDRBRCA = 0.00198, FDRTHCA = \(3.96\times ^\); Hyper-MS2: FDRBRCA = 0.00396, FDRTHCA = \(2.43\times ^\); Additional file 1: Fig. S8A, B).
Next, we examined the signature’s predictive power for survival. We performed survival analysis using the same patient groups based on signature activities in individual cancer types where the specific signatures were present (Fig. 3A and Additional file 1: Fig. S9A). As a result, we found that Hypo-MS4 was strongly predictive of poor OS in 3 out of the 6 cancer types in which Hypo-MS4 was present, such as LIHC (HR = 1.51, P = 0.03), KIRC (HR = 2.11, P = 0.04), and KIRP (HR = 1.93, P = 0.04) (Fig. 3A). Moreover, Hyper-MS1 predicted better survival in 3 out of the 8 cancer types tested, such as KIRC (HR = 2.19, P = 0.02), KIRP (HR = 6.37, P < 0.0001), and THCA (HR = 12.79, P = 1 \(\times ^\)) (Fig. 3A). Since Hypo-MS4 is independent of age, our data suggest that Hypo-MS4 is a predictor of poor prognosis in multiple cancer types.
Fig. 3
Methylation signature activity associated with disease outcome. A Survival analysis for patients with different DNA methylation signatures was conducted for the individual cancer types in which the corresponding signatures were present. Patients were stratified into two groups based on median signature activity. B Relationships between cancer stage and the activity of methylation signatures in individual cancer types, where the corresponding methylation signatures are present. Patients were stratified into two groups according to tumor stage (stage I, II: pri.; stage III, IV: adv.)
Moreover, half of the methylation signatures (5/10) were related to cancer stage (Fig. 3B and Additional file 1: Fig. S9B). Our data showed that Hyper-MS1, Hyper-MS2, and Hypo-MS4 were significantly associated with advanced (stage III–IV) tumors (P < 0.05, Wilcoxon’s rank sum test; Fig. 3B). Among them, Hyper-MS1 was consistently associated with advanced cancer stage in four cancer types tested, namely, PRAD, THCA, KIRC, and KIRP, suggesting a unique role in tumor progression.
Taken together, these findings indicate that methylation signatures are associated with several clinical features. In particular, Hypo-MS4 activity is strongly correlated with poor patient survival, which is independent of age.
Methylation signatures associated with the tumor immune microenvironment (TIME)As previous studies demonstrated that DNA methylation is associated with cancer immunity [53, 54], we assessed the impacts of methylation signatures on the tumor microenvironment using known lymphocyte activities and immune cell fractions in nine cancer types [55, 56] and calculated the meta-effects (Spearman’s correlation coefficient, Methods) for each signature. Among the ten methylation signatures, four demonstrated significant correlations with the activities of lymphocytes in the tumor microenvironment (FDR < 0.05) (Additional file 1: Fig. S10A). In particular, Hypo-MS4 was strongly positively associated with diverse immune cell activities in most cancer types, such as CD4 + regulatory T cells (Spearman’s R > 0.3, P \(\le\) \(2.4\times ^\) in LUAD, BRCA, and HNSC) (Fig. 4A and Additional file 1: Fig. S10C) and lymphocyte infiltration (Spearman’s R > 0.3, P \(\le 1.4\times ^\) in LIHC, BRCA, HNSC, THCA, and KIRP) (Additional file 1: Fig. S10D).
Fig. 4
The relationship between hypomethylation signature 4 activity and the tumor immune microenvironment. A Correlations between the immune activity and CD4 + regulatory T cell infiltration and Hypo-MS4 activity in LUAD, BRCA and HNSC. B Correlation between the gene expression levels of CD274 and Hypo-MS4 activity in LUAD and HNSC. C Correlations between the gene expression levels of PDCDELG2 and Hypo-MS4 activity in LIHC, BRCA, HNSC, and KIRP. D Correlation between the gene expression levels of IDO1 and Hypo-MS4 activity in BRCA and HNSC. E Significantly enriched Reactome pathways of Hypo-MS4-associated genes (Q value < 0.05)
We further examined the correlation between the signature activity and the expression levels of immune-related genes and obtained consistent results for Hypo-MS4, Hypo-MS1, and Hypo-MS7 (Additional file 1, Fig. S10B). In particular, Hypo-MS4 activity was positively associated with the expression levels of immune checkpoint genes, namely, CD274, PDCD1LG2, and IDO1, in multiple cancer types (P ≤ 0.01; Fig. 4B-D; Additional file 1: Fig. S10E-G). Moreover, Hypo-MS4-associated genes were strongly enriched in immune pathways (Methods, Fig. 4E).
Taken together, the effects of Hypo-MS4 on the TIME are the most prominent among all the methylation signatures, suggesting its unique role in the interaction between cancer cells and the microenvironment and its potential influence on cancer immunity.
Hypo-MS4 is associated with tumor immune evasion and the ICI responseWe think that the somatic mutation burden or neoantigen load may underlie the association between Hypo-MS4 and cancer immunity. We analyzed the tumor mutational burden (TMB) and neoantigen load in the tumor types in which Hypo-MS4 was present. We observed no significant correlations between TMB and Hypo-MS4 in LUAD, LIHC, and KIRC and a moderate negative association in HNSC and KIRP (P < 0.05). In BRCA, TMB was positively associated with Hypo-MS4 (Additional file 1: Fig. S11A, B).
For the neoantigen load, we found that tumors with high Hypo-MS4 activity showed consistently lower neoantigen loads in 4 out of the 6 cancer types tested, including LIHC (P = 0.024), HNSC (P = 0.0043), KIRC (P = 0.54), and KIRP (P = 0.037) (Fig. 5A). In LUAD patients, there was no significant association between neoantigen load and Hypo-MS4 activity (P = 0.96; Additional file 1: Fig. S11C). In BRCA, the neoantigen load was positively associated with Hypo-MS4 status (P = 0.00031; Additional file 1: Fig. S11D). However, this association with neoantigen load became nonsignificant within BRCA patients with distinct estrogen receptor (ER) statuses (Additional file 1: Fig. S11D).
Fig. 5
Hypo-MS4 is a signature of tumor immune responses that is associated with distant metastasis. A Comparison of neoantigen load between tumors of low and high Hypo-MS4 activity in four cancer types. B Correlation analysis of the fraction of cancer cells with high Hypo-MS4 activity and the fraction of CD4 + regulatory T cells in melanoma. C Fractions of cancer cells with high Hypo-MS4-GES scores in primary lesions and lymph node metastases from patients with colorectal cancer in single-cell methylome datasets of three patients, CRC01, CRC02, and CRC10. D Differences in Hypo-MS4 activity in responders and nonresponders to ICI therapy (Wilcoxon’s rank sum test) according to two methylome sequencing studies of melanoma patients. E Hypo-MS4-GES score in responders and nonresponders to ICI therapy (Wilcoxon’s rank sum test), in patients with gastric cancer (Kim et al. (2018) [45]) and in patients with melanoma (Liu et al. (2019) [46]; Riaz et al. (2017) [47]). F Fraction of Hypo-MS4-positive cells in ICI therapy responders and nonresponders (Wilcoxon’s rank sum test) before and after ICI treatment based on the single-cell transcriptome of melanoma
To further verify the interactions between Hypo-MS4 and the tumor immune microenvironment, we resorted to a dataset with matched single-cell transcriptomic and methylomic profiles from colorectal cancer (CRC) [57]. Our data showed that the fractions of cancer cells with high Hypo-MS4 activity were consistently enriched in lymph node metastases and liver metastases compared with those in primary tumors (Fig. 5B). To leverage other transcriptome datasets of the tumor immune microenvironment and ICI treatments where methylome information is unavailable, we derived a 156-gene expression signature (GES) for Hypo-MS4 by correlating single-cell gene expression levels to Hypo-MS4 activity (AUC = 0.88; Additional file 1: Supplementary Methods; Additional file 1: Fig. S11E) [57]. With single-cell RNA-Seq data from HNSC, we observed the same positive correlation between the fraction of cancer cells with high Hypo-MS4-GES scores and lymph node metastasis [58] (Additional file 1: Fig. S11F). Moreover, in another study of single-cell transcriptome data from colorectal cancer in which tumor-infiltrating lymphocytes were profiled along with tumor cells [59], our data showed that the fraction of cancer cells with a high Hypo-MS4-GES was positively correlated with the fraction of regulatory CD4 + T cells (Pearson’s R = 0.46, P = 0.028; Fig. 5C) and negatively correlated with the fraction of tumor-infiltrating CD8+ T cells (Pearson’s R = -0.36, P = 0.087) (Additional file 1: Fig. S11G), both of which promote immune evasion of tumor cells [60, 61].
Prompted by these findings, we examined whether Hypo-MS4 was predictive of the response to immune checkpoint inhibitors (ICIs). To this end, we analyzed Hypo-MS4 activity in a dataset of advanced metastatic melanoma patients who received anti-PD1 therapy (n = 43), in which DNA methylation was profiled prior to therapy [43]. We found that the activity of Hypo-MS4 was greater in the nonresponders to the PD-1 inhibitor than in the responders (Wilcoxon’s rank sum test, P = 0.038) (Fig. 5D, left panel). In another cohort of melanoma patients who received anti-CTLA4 or anti-CTLA4 + anti-PD1 therapy (n = 35) [44], Hypo-MS4 was consistently associated with poor responses but was less significant (Fig. 5D, right panel, P = 0.36). We then compared Hypo-MS4 activity between ICI responders and nonresponders among patients with melanoma before treatment based on bulk gene expression profiles [45,46,47]. As a result, Hypo-MS4-GES scores were consistently greater in nonresponders than in responders to ICI treatment in all three cohorts (gastric cancer, Kim et al. 2018, P = 0.025; melanoma, Liu et al. 2019, P = 0.019; melanoma, Riaz et al.2017, P = 0.51; Wilcoxon’s rank sum test) (Fig. 5E). Finally, in a single-cell RNA-seq dataset of melanoma [48], our data consistently showed that the fraction of cancer cells with high Hypo-MS4-GES scores was greater in nonresponders to ICI before treatment (baseline, P = 0.156; Wilcoxon rank-sum test, Fig. 5F) and further increased after treatment (posttreatment, P = 0.013, Wilcoxon rank-sum test), suggesting that Hypo-MS4-GES is strongly predictive of resistance to ICI therapy.
In summary, Hypo-MS4 is strongly associated with biomarkers of known immune processes in the tumor microenvironment. In particular, these associations are coupled with the consistent predictive power of Hypo-MS4 for poor response to immunotherapy in multiple cancer types.
Determinants of DNA methylation signaturesInspired by recent advances in cancer epigenetic reprogramming and our previous findings [39], we believe that revealing the underlying biological processes of methylation signatures is important. Here, we performed an integrated analysis to identify possible deterministic genes of the methylation signatures (Methods).
First, we used mutational status as an instrumental variable (IV) to identify genes whose expression levels influenced the methylation signatures (dependent variable; Methods) [62, 63]. As a result, we identified eleven deterministic gene mutations (six frameshift mutations and five missense mutations; Table 1), which significantly affected the activity of the methylation signatures (FDR < 0.05) (Table 1). Notably, Hypo-MS4 activity is associated with both TP53 and FOXA1 (Table 1). We further investigated whether the deterministic genes bind specifically to the DMPs of certain methylation signatures (Methods). As a result, the FOXA1-binding motifs were significantly enriched in Hypo-MS4-related DMPs in the Panc1 cell line (FDR < 0.05; Table 1) [64]. In addition, there were other genes in which the DNA-binding motifs were significantly enriched in the DMPs related to specific methylation signatures, such as NRF2 (Hypo-MS1), TEAD1 and TEAD3 (Hyper-MS2), RUNX1 (Hypo-MS1, Hypo-MS3), and RUNX2 (Hypo-MS1, Hypo-MS3, Hypo-MS3) (FDR < 0.1, Table 1).
Table 1 Instrumental variable regression analysis and motif analysis to determine the deterministic genes of the DNA methylation signaturesOur data strongly suggest that the activities of the methylation signatures are determined by the interaction of cancer driver mutations and genes with specific DNA-binding activities.
Hypo-MS4 surrogates for methylation at FOXA1-binding sites and dependent on TP53 mutationOur data showed that the activity of Hypo-MS4 is strongly influenced by TP53 and FOXA1. We retrieved the FOXA1-binding landscape for the DMPs related to Hypo-MS4 in multiple cancer cell lines (A549, HepG2, T47D and MCF-7) (Fig. 6A and Additional file 1: Fig. S12A, B) and tumor tissues (BRCA and COAD) (Fig. 6B) and showed that the FOXA1-binding affinity selectively peaked at the center of the DMPs. Moreover, the mean methylation levels of FOXA1-binding sites were strongly correlated with the activity of Hypo-MS4 in both TCGA (Additional file 1: Fig. S12C) and single-cell cohorts (Fig. 6C). On the other hand, we did not find any FOXA1-binding affinity at Hypo-MS4-down-DMPs (Hypo-MS4 based on 1-β) (Methods, Additional file 1: Fig. S12D).
Fig. 6
Correlation between deterministic genes and Hypo-MS4 activity. A FOXA1 binding landscape of Hypo-MS4-related DMPs in the gene body (n = 162) and TSS regions (n = 98) in the A549 and HepG2 cell lines. B FOXA1 binding landscape of Hypo-MS4-related DMPs in the gene body (n = 162) and TSS regions (n = 98) in colorectal and breast cancer tissues. C Correlation between the mean methylation level of FOXA1-binding sites and Hypo-MS4 activity in single-cell methylome data of colorectal cancer patients (left panel: FOXA1-binding sites in the TSS; right panel: FOXA1-binding sites in the gene body). D FOXA1 expression levels in patients with low and high Hypo-MS4 activity (according to the median Hypo-MS4 activity) in TP53-MUT and TP-53 WT tumors from LIHC, BRCA, and HNSC. E Hypo-MS4-GES scores increase in TP53-MUT and TP53-WT melanoma patients who are nonresponders to immune therapy
We further verified the impact of the TP53 mutational status on Hypo-MS4 activity in cancer types in which both Hypo-MS4 and TP53 mutations were present, with a sample size greater than 10. The results revealed that Hypo-MS4 activity increased with increasing TP53 mutation in 3 out of 4 cancer types, namely, LUAD (P = 0.81), LIHC (P = 0.069), and BRCA (P < 2.22 \(\times ^\)) (Additional file 1, Fig. S12 E, F). In addition, we conducted an analysis of the most commonly recognized hotspot mutations in TP53, namely, R175H, R248Q, R273H, Y220C, and R249S. However, due to the limited number of observations within each TP53 mutant group, we were only able to detect a significant increase in Hypo-MS4 activity within the TP53 (R175H) group in BRCA compared to that in the TP53-WT group (Additional file 1: Fig. S12G).
Next, we sought to evaluate the interactive effects of TP53 and FOXA1 on Hypo-MS4 activity. We observed a negative association between Hypo-MS4 activity and FOXA1 expression in the TP53-MUT group in 3 out of the 4 mentioned cancer types (LIHC: P = 0.086; BRCA: P = 9.7 \(\times ^\); HNSC: P = 0.011; Wilcoxon rank-sum test, Fig. 6D and Additional file 1: Fig. S13A). However, in the TP53-WT group, the effect was significant only for BRCA cells, and the effect also interacted with ER status (Fig. 6D, Additional file 1: Fig. S13B). In addition, we performed multilinear regression analysis (Methods) to further validate the interaction effect of FOXA1 and TP53 on Hypo-MS4 activity. The interaction between FOXA1 expression and TP53 mutational status was found to be a significant predictor of decreased Hypo-MS4 activity in 3 out of 4 cancer types (LIHC: coeff = − 97.68, P = 9.4 \(\times ^\); BRCA: coeff = − 54.40, P = 0.028; HNSC: coeff = − 81, P = 0.03; and LUAD: coeff = 7.6, P = 0.8; Additional file 1: Fig. S13C). Our results suggest that Hypo-MS4 activity in cancer cells is determined by TP53 mutation, FOXA1 expression, and their interaction. FOXA1 can demethylate its binding sites in a POLB-dependent manner [65]. Our observation of TP53 mutant tumors consistently showed that suppression of FOXA1 resulted in reestablishment of methylation at Hypo-MS4-DMPs (FOXA1 binding sites), hence increasing Hypo-MS4 activity.
We further investigated whether the predictive power of Hypo-MS4 is influenced by the TP53 mutational status and FOXA1 expression. In LIHC, KIRC, and KIRP, Hypo-MS4 showed no significant association with OS in patients with TP53-WT or TP53-MUT tumors, suggesting that the effect of Hypo-MS4 on survival is mainly driven by the mutational status of TP53 (Fig. 3A, Additional file 1: Fig. S14A). The same effects were observed within high- and low-FOXA1-expressing tumors (Additional file 1: Fig. S14B). Nevertheless, we observed a consistent increase in Hypo-MS4 activity in melanoma patients who had a poor response to ICIs [46], within the TP53-WT (P = 0.031) or TP53-MUT (P = 0. 28, Fig. 6E). The effect remained significant after we adjusted for TP53 mutational status and FOXA1 expression in a multivariate logistic regression. (ORHypo-MS4 = 0.74, P = 0.02; Additional file 1: Fig. S14C; Methods). Furthermore, neither the mutational status of TP53 nor FOXA1 expression alone predicted an ICI response (Additional file 1: Fig. S14D, E). These findings imply that Hypo-MS4 could ser
留言 (0)