
記住我



This study establishes that inflammatory signaling via the CD40L–CD40–TRAF6 axis plays a crucial role in diabetic (db/db) and hypertensive (ATII) mice. We also demonstrate that TRAF6 inhibition has beneficial effects in these animal models. In addition, we show that some markers (like CD40L, NOX2, and 3NT) identified in the mouse models showed a pattern of step-wise upregulation in CHD patients by the presence of comorbidities such as hypertension or hypertension + diabetes. CD40L and CD40 were also significantly correlated with an appreciable number of targets of a low-grade inflammatory cluster in hypertensive/diabetic patients.
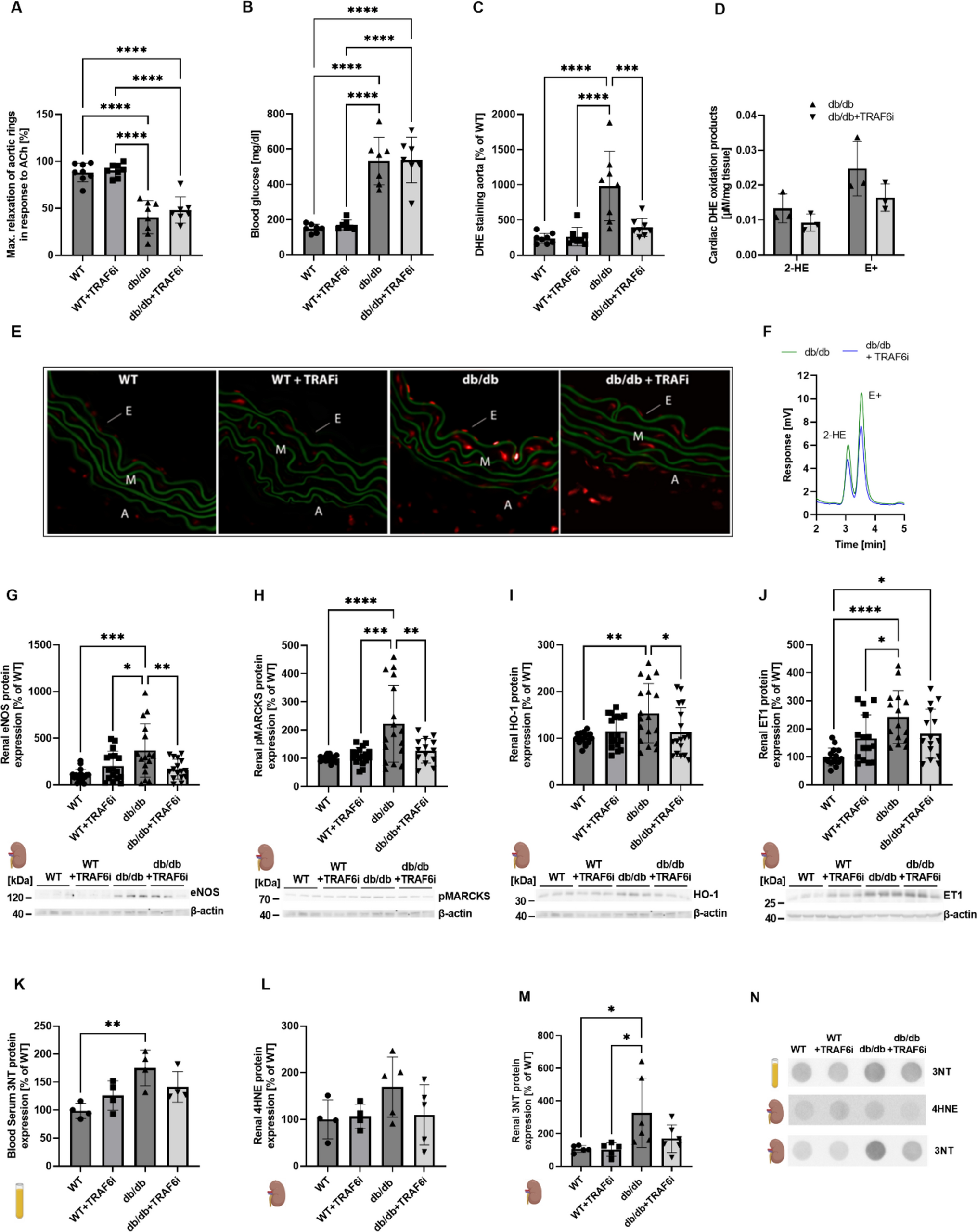
Animal dataA previous study showed that TRAF6 inhibition (often also termed “CD40-TRAF6” inhibition) by compound 6877002 suppressed the expression of TNFα, CCL2, and different interleukins in activated macrophages and diminished the expression of TNFα, CD40, and CD40L by trend in isolated B cells [37]. We showed that TRAF6 inhibition in diabetic mice (db/db), at least by trend, normalizes the upregulation of proteins centered around platelet activation (e.g., CD40L, TSP1, PAR1). Previously, we established that TRAF6 inhibition partially prevented endothelial dysfunction, weight gain, increase in HbA1c values, oxidative burst of whole blood leukocytes, cardiac RAGE signaling, vascular ROS formation, and impaired catalase/glutathione peroxidase 1 expression in db/db mice [38]. It is already known that the release of soluble CD40L and mainly CD40L–CD40 mediated signaling of platelets, endothelial cells, and monocytes strongly affects CHD [1, 13, 39]. Indeed, one way of expressing CD40 and mobilizing CD40 on the membrane in endothelial cells is induced via PAR-1, which could represent another link between thrombosis and inflammation [30]. Besides sCD40L, TSP-1 could be a potential biomarker in diagnosing acute coronary syndrome (ACS) [15]. We conclude from this data that inhibition of the CD40–TRAF6 signaling cascade in diabetic mice is suitable for suppressing thrombosis and the progression of CD40L–CD40-mediated inflammation.
Inhibition of TRAF6 also decreased the expression of proteins that are involved in reactive oxygen species formation (e.g., p47phox, NOX2), inflammatory signaling (e.g., VCAM-1, RAGE), and cell death (e.g., caspase-3) in diabetic animals. NADPH oxidase (NOX2) activation is associated with superoxide production, activation of NF-κB signaling, and release of pro-thrombotic molecules (e.g., sCD40L). Also, high CD40 surface protein levels affect the activation of NOX2 [32] [28]. In addition, RAGE could be activated via advanced glycation end products (AGE) to enhance the expression of adhesion and inflammatory molecules, including CD40 expression and mobilization to the surface of monocytes, which results in increased T cell activation and TNFα and interferon-γ levels [40]. With our present data, we could show that these processes are suppressed by TRAF6 inhibition in diabetic mice. Also, upregulated eNOS, reflecting compensatory upregulation of the uncoupled enzyme, was normalized by inhibition of the CD40–TRAF6 pathway in the heart and kidney. Furthermore, renal phosphorylation of MARCKS, reflecting p47phox-mediated NOX2 activation, and antioxidant response (e.g., HO-1) and vasoconstrictor expression (e.g., ET-1) was suppressed by TRAF6 inhibition in db/db mice. Also, aortic ROS formation was increased in diabetic mice, leading to endothelial dysfunction, all of which was improved, at least by trend, by TRAF6 inhibitor treatment. In contrast, the severely increased blood glucose levels in db/db mice were not decreased by TRAF6 inhibition.
Previous data indicate that ATII-mediated arterial hypertension is related to increased CD40L expression, most likely mediated by monocytes [43] and endothelial cells [19, 24]. Accordingly, we here observed a similar trend for the protective effects of TRAF6 inhibition in hypertensive mice as documented in diabetic mice. Indeed, in hypertensive mice with TRAF6 inhibitor treatment, cardiac RAGE and TXNIP expression were significantly reduced. TXNIP is a key component of high-mobility group box 1 protein (HMGB1) and NLR family pyrin domain containing 3 protein (NLRP3) activation, leading to inflammation. In addition, renal RAGE, HO-1, and VCAM-1 showed a similar trend in hypertensive mice. Oxidative stress markers such as 3NT, MDA, and 4HNE were upregulated in the blood or kidney of diabetic or hypertensive mice and normalized by CD40-TRAF6 inhibition. Of note, these biochemical changes were also mirrored by functional changes—TRAF6 inhibition partially normalized increased systolic blood pressure, endothelial dysfunction, and aortic ROS formation in hypertensive mice. Moreover, some of the mentioned biochemical markers (like CD40L, NOX2, and 3NT) identified in the mouse models were also step-wise upregulated in CHD patients by the presence of comorbidities such as hypertension or hypertension + diabetes.
Human protein dataIncreased cardiovascular risk is observed in patients suffering from systemic inflammatory diseases [6, 31]. Therefore, CVDs were classified as an inflammatory-related entity [3]. This correlation was also demonstrated in patients with ACS in which C-reactive protein (CRP) levels positively correlated with adverse clinical outcomes [33]. In the CANTOS trial, pharmacological agents like Canakinumab, an IL-1β antagonist, are employed in patients with high cardiovascular risk as anti-inflammatory therapy to improve their outcomes [34]. Besides inflammation, oxidative stress represents a hallmark of most CVDs, characterized by a mismatch between ROS formation and degradation [17, 20], e.g., as shown by a positive correlation between glutathione peroxidase-1 and outcomes in patients with CVD [8] as well as markers of oxidative stress and cardiovascular mortality [36].
In our proteomic analysis of CHD patients’ plasma, we identified some step-wise increased targets in association with the number of their comorbidities (CHD < CHD + HT < CHD + HT + T2DM). Cluster analysis identified TNFα as a central player. Detailed extended discussion of all regulated targets and the cluster data are provided in the online Supplemental Data file. Taken together, the central position of TNFα in the plasma protein cluster analysis provides evidence for an involvement of the CD40(L)-TRAF6 pathway in patients with HT and T2DM as comorbidities, which is further supported by the substantial correlations of CD40L and CD40 with a majority of other targets of the low-grade inflammatory cluster (Suppl. Tables S1 and S2, Figs. S3 and S4). Previous work showed that silencing of CD40 or TNF receptor (TNFR)-associated factor 6 (TRAF6) gene largely abrogated phosphorylation and nuclear translocation of NF-κB (p65), and hence TNFα expression [5]. Blockade of CD40 and CD40L interactions with neutralizing antibodies significantly reduced monocyte release of inflammatory mediators and migration by CCL2 (MCP-1). Another study reported upregulation of CD40, TRAF2, and TRAF6 in patients with diabetic retinopathy, where CD40 was associated with the expression of pro-inflammatory molecules such as intercellular adhesion molecule 1 (CD54), CCL2, and TNFα [42]. Several other factors identified in the cluster analysis of patients with HT and T2DM were previously reported for pathways associated with CD40L and other tumor necrosis factor superfamily ligands in HIV infection [23]. It is also well established that TNFα cooperates with CD40L, RANK, and IL1β to regulate TRAF6/NF-κB signaling [29], which is also supported by the observation that TRAF6 inhibition by compound 6877002 prevents CD40L-dependent activation (nuclear translocation) of NF-κB [7].
Human RNA sequencing dataIn accordance with our proteomic data, we found in CHD patients with comorbidities (CHD < CHD + HT + T2DM) increased aortic gene expression of NK cell-specific markers (e.g., CD244, KLRD1, NCR1), apoptotic markers (e.g., Gal1, TNFRSF21), cytokines/chemokines (e.g., TNF, CCL3, CCL4, IL12, IL8), T cell activation-associated markers (e.g., TNFRSF4, TNFRSF9, CD5, ICOSLG, CD70), and pro-angiogenic markers (e.g., AngPT1, TIE2).
The comparison of our proteomic data with our RNA-seq data (see Suppl. Table S8) showed only a small overlap in the regulated genes and differences in the regulation direction. In the RNA-seq analysis of the CHD + HT vs. CHD group, only CD8 and CD5 were upregulated, and CCL13 (MCP4) and CA9 (CAIX) were downregulated. There were no significant changes in the mRNA expression of the other genes. The comparative analysis of the proteomic data with the RNA-seq data of the comparison CHD + HT + T2DM vs. CHD shows more genes regulated on the mRNA expression level. However, in this comparison, several genes are downregulated on the mRNA level, which has been found to be upregulated on the protein level in the plasma. In this group 4 genes (PGF, AngPT1, CXCL8, NCR1) showed enhanced mRNA expression, whereas 5 genes (CA9, GAL, CXCL11, TNFRSF21, LAG3) showed reduced mRNA expression. However, as protein expression in the plasma (whole body) and mRNA expression in aorta samples are supposed only to show a minor overlap, our data are not surprising. Detailed extended discussion of IPA analysis of RNA-seq data is provided in the online Supplemental Data file, including suppl. Figs. S7,S8,S9.
The proteomic and transcriptomic data of human samples are further supported in this study by protein expression analysis. In patients’ plasma, we could observe the same trend of upregulation for proteins involved in reactive oxygen formation/oxidative stress (e.g., NOX-2, 3NT) and inflammation (e.g., CD40L, CD68), like in the proteomic analysis before (Suppl. Fig. S5).
Limitations of the studyThe chosen mouse model for diabetes (db/db) develops a quite severe vascular phenotype, as reported previously [38]. However, even a short-term TRAF6 inhibition partially reversed vascular complications such as impaired endothelial function, vascular ROS formation, and increased HbA1c and RAGE signaling [38]. Our mouse model of induced arterial hypertension via ATII treatment reflects only one possible pathway underlying the development of hypertension. In addition, the model of hypertension with one week of ATII infusion does not fully reflect hypertension-triggered end-organ damage by remodeling and accumulating oxidative modifications (e.g., in the kidney and heart). Accordingly, the impact of TRAF6 inhibition on end-organ damage could not be tested to a full extent here. However, we have previously shown that CD40L deficiency prevents all major complications induced by ATII treatment [19]. Hypertension development is multifactorial in humans, with the renin–angiotensin–aldosterone system playing a substantial role. Nevertheless, we think this model is suitable for our studies, especially in the context of the CD40L–CD40–TRAF signaling. We could show in the past that the ATII model is primarily driven by the immigration of (CD40L expressing) inflammatory cells. The negative influence of ATII-induced arterial hypertension on vascular function and severe blood pressure increase is well documented and has been intensively investigated, particularly in our research group [19, 43] and by others [14, 26]. Although the ATII-based hypertension mouse or rat model causes severe hypertension and drastic elevation of oxidative stress and inflammation, it is frequently used to study the pathophysiology of arterial hypertension in a short time window of 1–2 weeks. The beneficial action of all classical antihypertensive drugs, such as AT1-receptor blockers and angiotensin-converting enzyme inhibitors, supports the relevance of these models for human hypertension.
Concerning the Olink proteomic data, the variability within individuals arises from several factors aside from the differentiating morbidities/comorbidities. Circadian rhythms, environmental stressors, age, diet, and known or unknown factors can regulate the plasma proteome. The cohort size was too small to account for these confounding factors. They can be overcome by increasing the sample number. We quantified the plasma proteome of 88 individuals with a range of comorbidities. The number of patients in the individual cohorts is likely limiting; we also cannot rule out unexpected confounding factors. In addition, we monitored a small number of proteins in a targeted fashion, limiting the breadth of analysis.
Compared to RNA-Seq data obtained with inbred animals, there is a greater heterogeneity in the transcriptomes of tissue samples obtained from human beings. These heterogeneities are likely related to differences in the DNA sequences of the different participants and, therefore, genetically related differences in the transcriptional activity. This fact results in lower p-levels in gene expression compared to healthy and diseased participants. This higher variance may result in undetected disease-related transcriptomic changes. To circumvent the limitations of the RNA-Seq method, larger numbers of healthy/ill tissues are needed. However, the obtainment of healthy aorta samples is quite problematic. The analysis of our RNA-Seq data showed relatively high heterogeneity in the mRNA expression in samples of the participants of one group. However, for example, the IPA analyses of the CHD + HT vs. CHD comparison resulted in detecting the CD40L–CD40 pathway as a major regulator of the gene expression changes, which fits quite well with our animal data.
Another limitation of our RNA-Seq studies is the usage of whole aorta specimens. As in this specimen, different cells (endothelial cells, smooth muscle cells, fibroblasts, immune cells, etc.) are included; the specific gene expression cannot be related to a specific cell type. In addition, infections may change the number of immune cells in the specimen and disturb the data.
As all participants in the study were treated with several drugs (partially overlapping between the different groups), the effects of the drugs on gene expression may also mask disease-related transcriptional changes.
Finally, since samples were used from a biobank, the included patients had no follow-up since this was not foreseen. Also, the sample size in two of the patient groups was quite low (n = 21 for CHD and n = 25 for HT + Dia), making the human study an exploratory pilot study. Also, the lack of the CHD patient group with only diabetes as a comorbidity represents a major limitation of the human part of the study.
Conclusion and clinical impactRecent large clinical trials demonstrated that anti-inflammatory therapy can reduce cardiovascular risk by broad suppression of inflammatory pathways, as shown by the COLCOT study [41], or by highly specific targeting of the inflammatory cascade, e.g., as shown by the CANTOS study [34, 35]. Our present study could link CD40L–CD40-mediated signaling and platelet activation, the activation of different leukocytes, oxidative stress, inflammation, and angiogenesis, thereby generating new hypotheses. The major pathomechanisms in hypertensive or diabetic mice are associated with CD40(L)–TRAF signaling. Also, in patients with CHD, some key mediators of this pathway were found (summarized in Fig. 6). We conclude that CD40L–CD40 signaling is important in mediating cardiovascular complications in mouse models of obesity, dyslipidemia, hyperglycemia, and arterial hypertension. In addition, the proteomic and genomic analysis in plasma and vascular bypass tissue of CHD patients without comorbidities or with the comorbidities hypertension and/or hypertension + diabetes identified some key mediators of CD40(L)–TRAF signaling, centered around TNFα. However, the present data cannot clarify whether the step-wise increase in levels of low-grade inflammation markers is mediated directly by the comorbidities hypertension and/or diabetes or by the more severe CHD/events driven by these comorbidities; however, the disease state characteristics did not change significantly by the comorbidities.
Fig. 6
Central scheme. Upper left panel: the central pathomechanisms associated with CD40(L)–TRAF signaling in hypertensive or diabetic mice. Upper right panel: protective effects of TRAF6i treatment in hypertensive or diabetic mice. Lower panel: markers associated with CD40(L)–TRAF signaling observed in CHD patients with no comorbidity or hypertension or hypertension + diabetes. It must be stressed that an appreciable part of the targets mentioned here were only changed by trend. WBC white blood cell, Tsp-1 thrombospondin-1, CD cluster of differentiation, CD40L CD40 ligand, PAR1 protease-activated receptor 1, VCAM-1 vascular cell adhesion molecule-1, TXNIP thioredoxin-interacting protein, NOX2 NADPH oxidase 2, RAGE receptor for advanced-glycation-end-products, TNF tumor necrosis factor, TRAF6i TNF receptor associated factor-6 inhibitor, CHD coronary heart disease, IL12 interleukin-12, TNFRSF4 (or ox40 or CD134) TNF receptor superfamily member-4, TNFRSF9 (or 4-1BB or CD137) TNF receptor superfamily member-9, ICOSL inducible costimulator ligand, AngPT1 angiopoietin-1, PGF placental growth factor, TIE2 angiopoietin receptor-2, CCL3/CCL4 C–C motif chemokine ligand-3/-4, CXCL11 C-X-C motif chemokine ligand-11, 3NT 3-nitrotyrosine, KLRD1 killer cell lectin like receptor D-1, NCR1 natural cytotoxicity triggering receptor-1, DR6 death receptor-6, Gal1 galactin-1, NK natural killer cell. Created with BioRender.com
In conclusion, hypertension and lipid metabolic disorders are important risk factors and leading causes of atherothrombotic vascular diseases like MI and cerebral ischemia. A targeted CD40-TRAF6 inhibitor treatment could provide a novel therapeutic strategy for these diseases, which circumvents the potential risks of a general blockade of CD40L or CD40-like thromboembolic events and solid immune suppression. It remains to be established if a long-term CD40-TRAF6 inhibitor treatment also provides some risks, like the blockade of CD40L or CD40. Finally, human studies are needed to test the beneficial effects of TRAF6 inhibition in hypertension and diabetes.
留言 (0)