
記住我

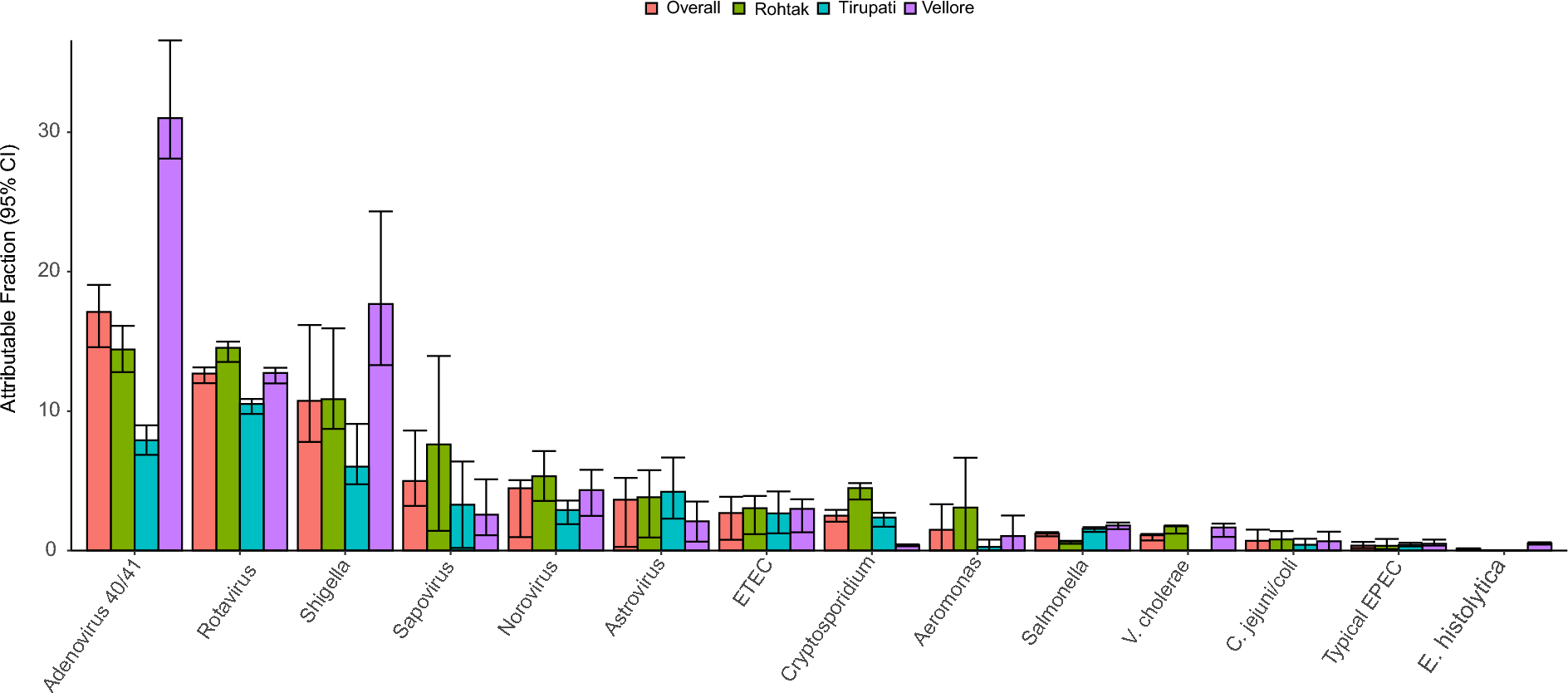

First, the source of the data (own vs. public) were analyzed by PCoA plot (Fig. 1A). Then, 1102 samples were clustered according to the relative abundance of bacteria at the genus level using the JSD distance metric. Considering the silhouette width, CH index, DBI index, and Dunn index, three clusters are determined when K = 3 (Fig. 1B). Based on the dominant bacteria in each group, the three enterotypes were respectively designated Streptococcus (S_E, n = 390), Bacteroide (BA_E, n = 452), and Blautia (BL_E, n = 260). The eight most abundant genera in each enterotype are shown in Fig. 1C. In brief, Streptococcus (23.02%) and Ruminococcus (14.52%) were relatively abundant in S_E type. Bacteroides (46.51%) and Blautia (13.91%) were relatively abundant in BA_E type; Blautia (35.87%) and Coprococcus (15.50%)were relatively abundant in BL_E group. A PCoA plot confirmed the differences among the three enterotypes, while the BA_E and BL_E enterotypes showed a certain degree of overlap (Fig. 1D). In addition, we observed the number of people with different health states in each enterotype (Fig. 1E). The S_E group included 106 healthy, 60 adenoma, and 224 CRC subjects; the BA_E group included 165 healthy, 168 adenoma, and 119 CRC subjects; the BL_E group included 96 healthy, 92 adenoma, and 72 CRC subjects. Thus, compared with the BA_E and BL_E enterotypes, the S_E group comprised the highest proportion of patients with CRC (57% vs. 26% and 28%). The proportions of adenoma and healthy subjects with the BA_E and BL_E enterotypes were essentially the same (35–37%) and somewhat higher than those of the patients with CRC (26–28%). Furthermore, the distribution of clinical factors such as age, sex, and BMI among three enterotypes was also analyzed. The results showed that there were no statistically significant differences in these clinical factors between the three enterotypes (P > 0.05) (Fig. 1F-H). Figure S1 shows the top eight bacterial genera in the three enterotypes for each of the subject populations, which indicates that even for subjects in the same population, there were notable differences among the three enterotypes with respect to gut microbiota composition.
Fig. 1
Gut enterotype analysis of 1102 samples. A: The source of the data (own vs. public) coloring by a simple PCoA plot. B: Calinski–Harabasz (CH) index analysis based on the Jensen Shannon divergence (JSD) distance. K = 3 is the optimal number of clusters. C: The top eight genera in each enterotype. D: Principal coordinates analysis (PCoA) plot of the three enterotypes. All samples (adenoma, n = 320; CRC, n = 415; healthy, n = 367) are clustered into “Streptococcus” (S_E, green), “Bacteroide” (BA_E, red), and “Blautia” (BL_E, blue) enterotypes. E: A bar chart showing the distribution of different disease states in three enterotypes. Red, green, and blue represent healthy control, adenoma, and blue CRC samples, respectively. F-H: The distribution of clinical factors such as sex, BMI and age among the three enterotype. ns meant no statistical difference between the two groups (P > 0.05)
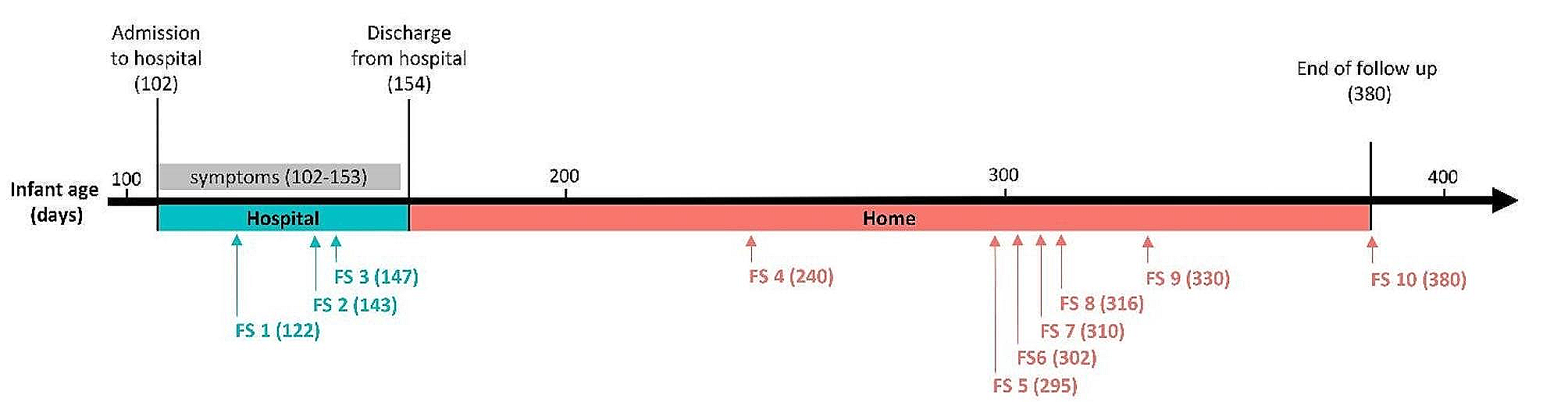
Gut microbiota composition in BA_E for three human cohorts at the genus levelIn the BA_E type, the bacterial composition at the genus level of the three human cohorts is shown in Fig. 2A (top 20 genera). Among the three groups, Akkermansia, Ruminococcus, Streptococcus, Gemmiger, and Subdoligranulum were identified as the five predominant genera. PCA plot indicated that the colony composition of the three populations was not significantly distinguished (Fig. 2B). In further analysis, LDA was used to screen for differences among the three subject populations with respect microbial community species, revealed 44 genera that differed among healthy, adenoma, and CRC subjects (Fig. 2C). Among these, eight genera, including Blautia, Faecalibacterium, and Lachnospira, were significantly enriched in the healthy group; 11 genera, including Coprococcus, Roseburia, and Alistipes, were significantly enriched in the adenoma group; 25 genera, including Fusobacterium, Oscillospira, and Porphyromonas, were significantly enriched in the CRC group.
Fig. 2
Distinct bacterial composition of samples from healthy subjects, adenoma patients, and colorectal cancer (CRC) patients in the BA_E enterotype. A: The community abundance of gut microbiota at the genus level. B: Principal component analysis (PCA) plot visualizing the three human cohorts. Red, green, and blue dots represent healthy control, adenoma, and CRC samples, respectively. C: Linear discriminant analysis (LDA) identified the differentially abundant genera among healthy, adenoma, and CRC samples
For each human cohort, we also examined correlations among the differential bacterial genera. In the healthy group (Figure S2A), Blautia showed negative correlation with Lachnospira (r = -0.044), while it was positively correlated with Selenomonas (r = 0.008). In the adenoma group (Figure S2B), Peptostreptococcus showed positive association with Parvimonas (r = 0.017) and negative association with Anaerostipes (r = -0.379). In the CRC group (Figure S2C), Leptotrichia was negatively correlated with Blautia (r = -0.225) and positive association with Raphanus (r = 0.211).
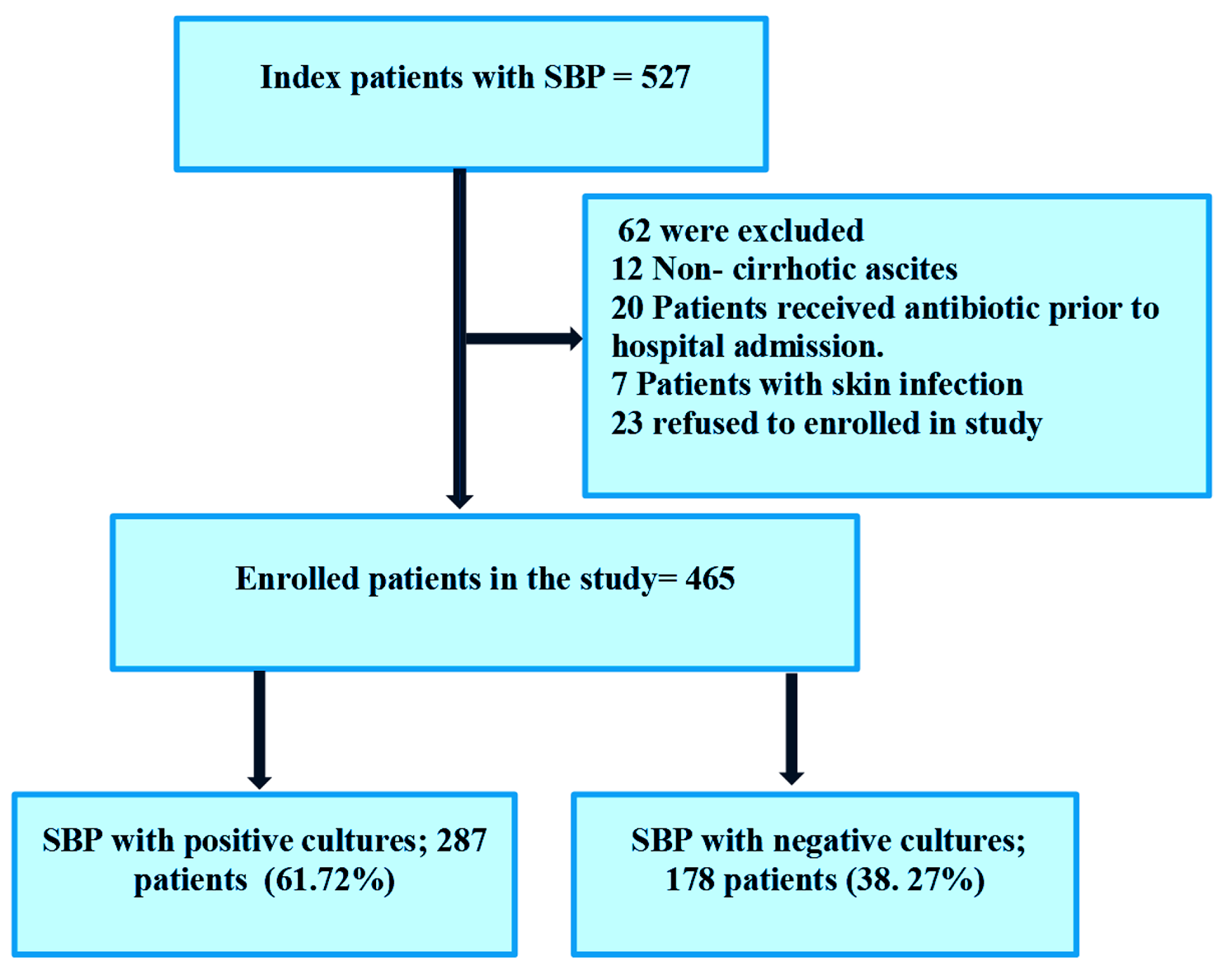
Gut microbiota composition in BL_E for three human cohorts at the genus levelIn the BL_E type, the relative abundances of the top 20 genera for three human cohorts are shown in Fig. 3A. In all three groups, Faecalibacterium, Roseburia, Bacteroides, Prevotella, and Dorea were the five predominant bacterial genera. PCoA plot showed that the three groups cannot be significantly separated (Fig. 3B). In addition, LDA method was employed to screen the specific genera for each group (Fig. 3C). Briefly, only one genus was significantly enriched in the healthy (Fusobacterium) and adenoma (Pseudomonas) groups. Eight genera were mainly identified in the CRC group, such as Collinsella, Porphyromonas, and Campylobacter.
Fig. 3
Distinct bacterial composition of samples from healthy subjects, adenoma patients, and CRC patients in the BL_E enterotype. A: The community abundance of gut microbiota at the genus level. B: PCA plot visualizing the three human cohorts. C: LDA identified the differentially abundant genera among the healthy, adenoma, and CRC samples
Next, we explored the correlation between differential gut microbiota of healthy, adenoma, and CRC samples, respectively. In the healthy group, Collinsella exhibited significantly positive correlation with Peptostreptococcus (r = 0.391), while it was negatively correlated with Pseudomonas (r = -0.822) (Figure S3A). In the adenoma group, Collinsella showed a significantly positive association with Parvimonas (r = 1), while it was negatively correlated with Pseudomonas (r = -0.883) and Fusobacterium (r = -0.857) (Figure S3B). In the CRC group, Gemella had strong positive correlation with Campylobacter (r = 1), and Peptostreptococcus had negative correlation with Pseudomonas (r = -0.578) (Figure S3C).
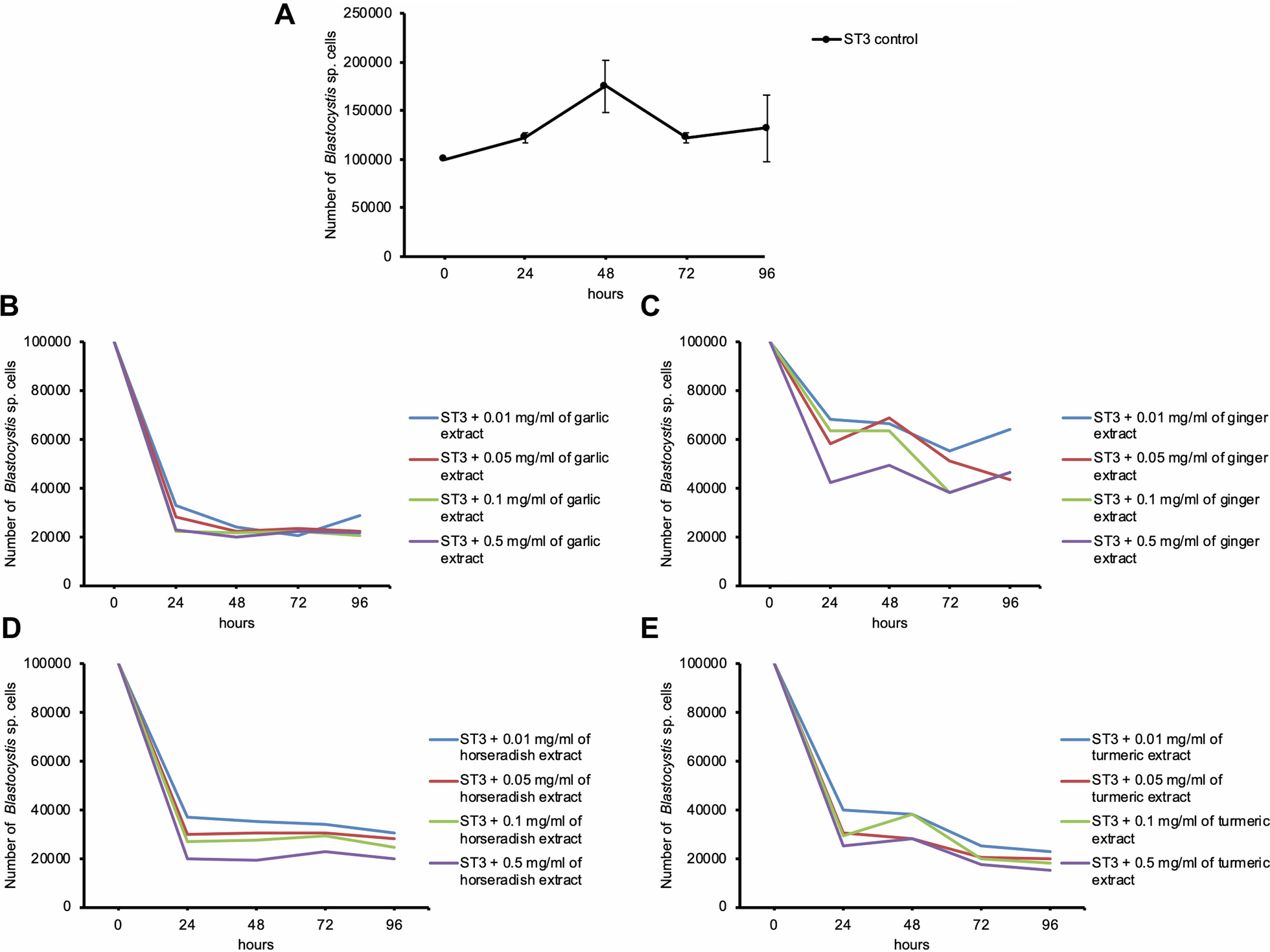
Gut microbiota composition in S_E for three human cohorts at the genus levelFurther, the composition of gut microbiota at the genus level in the S_E type was analyzed to describe specific changes in gut microbiota in different disease groups (Fig. 4A). For each of these populations, Bacteroides, Sporobacter, Gemmiger, and Succinispira were identified as the predominant bacterial genera. Compared with the healthy and adenoma groups, we observed higher relative abundances of Gemmiger, Clostridium, and Anaerosinus in CRC group patients In contrast, CRC group patients were characterized by the lowest relative abundance of Escherichia species, the abundances of which were notably higher in the adenoma group. According to the PCA plot, there was no significant structural differences in gut microbiota among the three groups, while a trend of segregation was observed between adenoma and CRC (Fig. 4B). LDA revealed a total of 78 predominant genera among the three groups, of which 13, 45, and 20 were detected in the healthy, adenoma, and CRC groups respectively (Fig. 4C). Among these, the genera Faecalibacterium, Bacteroides, and Roseburia were identified as dominant bacteria in healthy group; Escherichia, Raphanus, and Sneathia predominated the bacterial community in the adenoma group; Streptococcus, Lactobacillus, and Bifidobacterium were among the predominant genera in the CRC group.
Fig. 4
Distinct bacterial composition of samples from healthy subjects, adenoma patients, and CRC patients in the S_E enterotype. A: The community abundance of gut microbiota at the genus level. B: PCA plot visualizing the three human cohorts. C: LDA identified the differentially abundant genera among healthy, adenoma, and CRC samples
Next, we explored the correlation between 46 differential gut microbiota of healthy, adenoma, and CRC samples, respectively. In the healthy group, Faecalibacterium was negatively correlated with Peptostreptococcus (r = -0.786), while it was positively correlated with Roseburia (r = 0.366) (Figure S4A). In the adenoma group, Streptococcus showed positively correlation with Pseudomonas (r = 0.4), while it was negatively correlated with Bifidobacterium (r = -0.8) (Figure S4B). In the CRC group, Ruminococcus was found to be significantly negatively correlated with Bacteroides ( r = -0.335) and Actinomyces (r = -0.489). Alistipes was also found to have a strong positive relationship with Haemophilus and Abiotrophia (all r = 1), while it was negatively with Actinomyces (r = -0.587) (Figure S4C).
To verify the classification criteria of the three enterotypes, we also analyzed all samples in different disease states without performing enterotype. The results revealed that the three subject groups differed with respect to 89 genera of gut microbiota (Fig. 5A). Briefly, 17 bacterial genera, including Bacteroides, Faecalibacterium, and Fusobacterium, were significantly enriched in the healthy group, whereas 30 bacterial biomarkers, including Blautia, Coprococcus, and Escherichia, were significantly enriched in the adenoma group, and 42 biomarkers, including Streptococcus, Lactobacillus, and Dorea, showed highest abundance in the CRC group. In addition, different species of each enterotypes were screened through Venn analysis and LDA analysis. The results showed that there were 21 specific genera (Vibrio, Turicibacter, Sporobacter and etc.) in all samples, 10 specific genera (Treponema, Peptoniphilus, Mogibactenum and etc.) in BA_Eenterotypes, 2 specific genera (Epulopiscium and Colinsella) in BL_E enterotypes and 24 specific genera (Succinivibrio, Sporobacter, Dietzia and etc.) in S_E group, respectively (Fig. 5B-C). Moreover, Pseudomonas, Fusobacterium, and Peptostreptococcus, both in the three enterotypes and all samples (Fig. 5B and C), Among these, Peptostreptococcus was identified as a biomarker for CRC patients in the three enterotypes and all samples.
Fig. 5
The difference in gut microbiota profiles among healthy subjects, adenoma patients, and CRC patients based on all samples. A: LDA identified the differentially abundant genera among healthy, adenoma, and CRC. Red, green, and blue represent healthy control, adenoma, and blue CRC samples, respectively. B: A Venn diagram showing the overlap of microbiota within three enterotypes and all samples. Blue, green, and red indicate the BL_E, BA_E, and S_E enterotypes, respectively, and gray indicates all samples. C: Different species of each enterotypes screened by LDA analysis
Differential bacterial biomarkers in each enterotype can be used to distinguish three human cohorts based random forest classificationGiven our findings of different compositions of the three enterotypes in subject populations, we proceeded to establish whether these enterotypes have potential utility in differentiating among healthy, adenoma, and CRC subjects. Initially, we assessed the predictive ability of three-class classification in identifying healthy, adenoma, and CRC subjects. With respect to the BA_E group, the AUC of classification was 0.75 (F1 score = 0.54), with a sensitivity and specificity of 0.53 and 0.75, respectively (Fig. 6A). Furthermore, using this model, the characteristics of Peptostreptococcus, Porphyromonas, Parvimonas, Anaerococcus, and Coprococcus were found to have high importance scores. For the BL_E group, the AUC value was 0.62 (F1 score = 0.43), with a sensitivity and specificity of 0.43 and 0.71, respectively (Fig. 6B), and the top genera ranked in terms of importance were Epulopiscium, Porphyromonas, Pseudomonas, Peptostreptococcus, and Collinsella. For the S_E group, we obtained AUC, sensitivity, and specificity values of 0.78 (F1 score = 0.58), 0.56, and 0.8, respectively (Fig. 6C), and the genera Faecalibacterium, Pseudomonas, Raphanus, Bacteroides, and Streptococcus were assigned high importance scores. On the basis of these findings, we established that the predictive performance of S_E was superior to that of BA_E and BL_E. In addition, we also determined the predictive performance of differential bacteria without initial enterotype clustering (Fig. 6D). Using this model, we obtained an AUC value of 0.75 (F1 score = 0.55) for the classification of healthy, adenoma, and CRC subjects, with corresponding sensitivity and specificity values of 0.55 and 0.78, respectively, with Peptostreptococcus, Faecalibacterium, Pseudomonas, Blautia, and Porphyromonas being identified as the top ranked characteristic bacterial genera (See Fig. 7).
Fig. 6
Construction of a classification model to distinguish among healthy, adenoma, and CRC based on enterotypes and all samples. A: Random forest classifier prediction of the top 20 characteristic bacteria in the BA_E enterotype of the three human cohorts. B: Random forest classifier prediction of top 20 characteristic bacteria in the BL_E enterotype of the three human cohorts. C: Random forest classifier prediction of the top 20 characteristic bacteria in the S_E enterotype of the three human cohorts. D: Random forest classifier prediction of the top 20 characteristic bacteria in the three human cohorts based on all samples
Fig. 7
Construction of a classification model to distinguish between non-colorectal cancer and colorectal cancer samples based on enterotype and all samples. A: Predictive ability of the BA_E enterotype in distinguishing between non-CRC and CRC samples. B: Predictive ability of the BL_E enterotype in distinguishing between non-CRC and CRC samples. C: Predictive ability of the S_E enterotype in distinguishing between non-CRC and CRC. D: Predictive ability of all samples in distinguishing between non-CRC and CRC samples
We also used a two-class classification model to distinguish CRC from non-CRC samples. Using this model, we obtained AUC values 0.69, 0.68, 0.79, and 0.78 for BA_E, BL_E, S_E, and all samples, respectively. Among the bacterial genera, we obtained high importance scores for Peptostreptococcus, Porphyromonas, Parvimonas, Fusobacterium, and Coprococcus in the BA_E model; Porphyromonas, Peptostreptococcus, Fusobacterium, Parvimonas, and Clostridium in the BL_E model; and Faecalibacterium, Pseudomonas, Raphanus, Streptococcus, and Bacteroides in the S_E model. Considering all models combined, Peptostreptococcus, Faecalibacterium, Pseudomonas, Porphyromonas, and Blautia were identified as the top five most important bacterial genera. Consistent with the findings obtained based on three-class classification analysis, we found that among three enterotypes, S_E showed the highest predictive performance. However, compared with our analysis based on all samples, we identified no significant advantages regarding the disease-predictive power of enterotypes. These findings were confirmed using the validation sets (Figure S5).
留言 (0)