
記住我




Human PBMCs were obtained in accordance with the Declaration of Helsinki and the Dutch rules with respect to the use of human materials from volunteer donors. Buffy coats were obtained from healthy anonymized donors after their written informed consent, as approved by Sanquin’s internal ethics board. Human peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque Plus (Cytiva) density gradient centrifugation (GE Healthcare), and the cells were cryopreserved until further use. DCs were isolated from HLA-A2+ donors, while the CD4+ and CD8+ T cells used in this study were used regardless of their HLA type and were not necessarily from the same donor.
Patient samplesSeven head and neck squamous cell carcinoma (HNSC) patients [71] from the N16IMC trial (ClincalTrials.gov identifier: NCT03003637) along with 4 MMRp and 3 MMRd colorectal cancer (CRC) patients were included in this study. All patients were treatment naive when the samples were obtained (Supplemental Table 8). Tumor tissues from HNSC patients were cut into pieces of approximately 1–2 mm3 and cryopreserved until flow cytometric analysis. Tumor tissues from CRC patients were cut into small fragments in a Petri dish and enzymatically digested with 1 mg/mL collagenase D (Roche) and 100 μg/mL DNase I (Merck) in 5 mL of Iscove’s modified Dulbecco’s medium (IMDM; Gibco) supplemented with fetal bovine serum (FBS; Sigma) for 30 min at 37 °C in gentleMACS C tubes (Miltenyi Biotec). During and after incubation, the cell suspensions were mechanically dissociated in a gentleMACS Dissociator (Miltenyi Biotec). The cell suspensions were filtered through 70-μm cell strainers (Corning), washed in IMDM, and cryopreserved until further use. For flow cytometric analysis, tumor pieces from HNSC patients were first enzymatically digested with Liberase TL research grade (250 mg/ml; Merck) for 20 min at 37 °C, passed through 70-μm cell strainers (Falcon) and centrifuged at 400 × g for 5 min. After removal of the supernatant, the pelleted cells were resuspended in red blood cell lysis buffer and incubated at RT for 2 min. For samples from CRC patients, single-cell suspensions were quickly thawed and washed in RPMI-1640 medium + 20% FBS. Cells from HNSC and CRC patients were incubated with BD GolgiPlug (1:1000, BD Biosciences) for 5 h before staining. For staining, cells were first incubated in DNase I (0.1 mg/ml) at RT for 10 min, stained for surface markers, and fixed and permeabilized with a FOXP3 Staining Buffer Set (Thermo Fisher Scientific) according to the manufacturer’s protocol. Finally, the cells were stained for intracellular markers before harvesting. The collection of patient samples was approved by the Medical Research Ethics Committee of the Netherlands Cancer Institute-Antoni van Leeuwenhoek Hospital (file# NL57794.031.16) and Leiden University Medical Center (protocol P15.282), and all patients provided written informed consent. All specimens were anonymized and handled according to the ethical guidelines described in the Code for Proper Secondary Use of Human Tissue in the Netherlands of the Dutch Federation of Medical Scientific Societies.
Fluorescence-activated cell sorting (FACS)For in vitro DC-T-cell coculture and NanoString nCounter gene expression analysis, PBMCs were directly used for FACS. CD19+ cells were depleted before sorting using CD19 magnetic MicroBeads (Miltenyi Biotec) according to the manufacturer’s protocol. Staining was performed at 4 °C for 45 min in flow cytometry staining buffer (BD Biosciences). Antibodies specific for the following proteins were used: from BioLegend, CD1c (PE-Cy7, clone L161), CD3 (BV510, clone OKT3), CD4 (FITC, clone OKT4), CD8 (Percp-cy5.5, clone SK1), CD11c (PE/AF700, clone Bu15/3.9), CD14 (ef450, clone M5E2), CD19 (BV510, clone HIB19), CD25 (PE, clone BC96), CD45RA (AF700, clone HI100), CD141 (BV421, clone M80), and HLA-DR (BV605, clone L243); from BD Biosciences, HLA-DR (APC-Cy7, clone G46-6); and from Miltenyi Biotec, CD141 (APC, clone REA674). A Near-IR Dead Cell Stain Kit (Invitrogen), a Zombie Red Fixable Viability Kit (BioLegend) or 7-aminoactinomycin D (7-AAD, eBioscience) was used to exclude dead cells. To prevent clumping of dead cells, DNase I (0.1 mg/ml) was added before sorting. Cell sorting was performed on a BD FACSAria III (BD Biosciences).
Flow cytometryCell surface staining: Staining was performed at 4 °C for 30 min in flow cytometry staining buffer. Antibodies specific for the following proteins were used at a 1:50 dilution unless mentioned otherwise: from BioLegend, CD3 (BV650, clone OKT3), CD4 (BV785/PE-Cy7, clone OKT4), CD8 (AF700/BUV805, clone SK1), CD11c (APC/Fire750, clone 3.9), CD11c (AF700, clone Bu15), CD14 (Spark Blue550, clone 63D3), CD19 (BV510, clone HIB19), CD25 (BV785, clone M-A251), CD27 (APC/Fire810, clone O323), CD40 (BV711, clone 5C3), CD40L (PE-Cy7, clone SA047C3), CD44 (PE, clone C44Mab-5), CD45 (Spark UV387, clone HI30), CD45RA (Spark NIR685, clone HI100), CD45RO (BV570, clone UCHL1), CD69 (BV510, clone FN50), CD70 (Percp-cy5.5, clone 113-16), CD80 (BV785, clone 2D10), CD83 (BV421, clone HB15e), CD86 (BV510, clone IT2.2), CD127 (PE/Fire700, clone A019D5, 1:30), CD137 (BV711, clone 4B4-1), CD141 (BV421, clone M80), CCR7 (BV711/Percp-cy5.5, clone G043H7), CXCR3 (PE-Cy7, clone G025H7), HLA-A2 (PE-Cy7, clone BB7.2), HLA-ABC (FITC, clone W6/32), HLA-DR (PE/Fire810, clone L243), HLA-DR (APC-Cy7, clone L243), HLA-DR.DP. DQ (PE-Cy7, clone Tu39), PD-L1 (BV711, clone 29E.2A3), and XCR1 (BV421, clone S15046E); from BD Biosciences, CD3 (BV480, clone UCHT1), CD27 (BV650, clone L128), CD45 (PE-CF594, clone HI30, 1:100), CD45RO (PE-CF594, clone UCHL1, 1:100), CD303 (BV650, clone V24-785), CD326/EPCAM (BUV737, clone EBA-1), PD-1 (PE-CF594, clone MIH4); from Miltenyi Biotec, CD141 (APC, clone REA674); and from ImmunoTools, CD8 (FITC, clone HIT8a). AF488- or APC-conjugated HLA-A2/MART-126-35 tetramers were added together with cell surface staining antibodies. A Near-IR Dead Cell Stain Kit, a Zombie Red Fixable Viability Kit, a Zombie UV Fixable Viability Kit (BioLegend) or 7-aminoactinomycin D (7-AAD) were used to discriminate between live and dead cells.
For intracellular staining, a protein transport inhibitor (BD GolgiPlug) (1:1000) was added to the culture, which was incubated for 3 h before the cells were stained with cell surface markers. After surface staining, the cells were fixed and permeabilized using a BD Cytofix/Cytoperm Kit (BD Biosciences) or a FOXP3 Staining Buffer Set according to the manufacturer’s protocol. For phosphoprotein detection, cells were fixed with BD Cytofix™ Fixation Buffer (BD Biosciences) at room temperature for 20 min and permeabilized with BD™ Phosflow Perm Buffer III (BD Bioscience) at 4 °C for 30 min. Then, the cells were washed with flow cytometry staining buffer before intracellular staining. Antibodies specific for the following proteins were used at a 1:50 dilution unless mentioned otherwise: from BioLegend, CD107α (BV650, clone H4A3), β2m (PE, clone A17082A), Ki-67 (BV605, clone Ki-67, 1:30), Granzyme B (PE, clone QA16A02), CXCL9 (PE, clone J1015E10), CXCL10 (PE, clone J034D6), IFNγ (PE-Cy7, clone B27); from BD Biosciences, CD40L (PE-CF594, clone TRAP1, 1:100); from Miltenyi Biotec, pan-IFNα (APC, clone LT27:295); from R&D Systems, SLC19A1 (APC, clone mouse IgG2A 890513, 1:30), ISG15 (PE, clone rat IgG2A); from Cell Signaling Technology, phospho-TBK1 (PE, clone D52C2, 1:100), phospho-IRF3 (AF647, clone E7J8G), phospho-STING (AF488, clone D8K6H); from Bioss, TAP1 and TAP2(rabbit polyclonal anti-human); and from Thermo Fisher Scientific, CXCL13 (APC, clone DSBCX13, 1:30), FOXP3 (PE-Cy5, clone PCH101), and IFNβ (AF488, clone A1), as well as goat anti-rabbit IgG(H+L) Alexa Fluor 488 (use 1:200) and goat anti-rabbit IgG(H+L) APC (use 1:300) as secondary antibodies. Specific staining was confirmed with the fluorescence minus one (FMO) control. The antibodies and dilutions used are listed in Supplementary Table 9. Flow cytometry was performed using a BD LSR FortessaTM (BD Biosciences) or Cytek Aurora spectral flow cytometer (Cytek Biosciences). The data were analyzed using FlowJoTM software version 10.8.1 (BD Biosciences) or OMIQ software (Dotmatics).
STING pathway stimulation/inhibitionNaive CD4+ T cells were flow sorted on the basis of the CD3+HLA-DR-CD4+CD25-/lowCD45RA+ phenotype and cultured for 48-72 h after plating at 0.5 × 106 cells/well in 96-well round bottom plates (BD Falcon) in RPMI-1640 medium supplemented with 10% FBS and Antibiotic–Antimycotic Solution (Sigma) in the presence of hIL-2, hIL-7 and hIL-15 (Miltenyi, each 10 ng/ml); additionally, monoclonal antibodies against CD3 and CD28 were added to activate CD4+ T cells. In addition, CD4+ T cells were treated with a STING inhibitor (H-151, 15 ng/ml; InvivoGen) or a STING agonist (2′3′-cGAMP, 15 μg/ml; InvivoGen) for the last 8 h of culture. Intracellular IFNβ and phosphoprotein detection was performed using flow cytometry.
Enzyme-linked immunosorbent assay (ELISA)Naive CD4+ T cells were plated at 0.5 × 106 cells/well (200 μl/well) in a 96-well round bottom plate and cultured in medium containing 10 ng/ml hIL-2, hIL-7 and hIL-15 (Miltenyi). Additionally, monoclonal antibodies against CD3 (0.1 μg/ml, clone CLB-T3/4.E, Sanquin) and CD28 (0.2 μg/ml, clone CLB-CD28/1, Sanquin) were added to generate activated CD4+ T cells. The supernatant was collected after 48 h of culture and frozen at −20 °C until analysis. A Human IFN-Beta ELISA Kit with high sensitivity (PBL Assay Science) was used according to the manufacturer’s instructions.
ImmunoblottingNaive CD4+ T cells were cultured in medium supplemented with 10 ng/ml hIL-2, hIL-7 and hIL-15 (Miltenyi). Additional monoclonal antibodies against CD3 (0.1 μg/ml, clone CLB-T3/4.E, Sanquin) and CD28 (0.2 μg/ml, clone CLB-CD28/1, Sanquin) were added to generate activated CD4+ T cells. During the last 8 h of culture, the CD4+ T cells were treated with a STING agonist (2′3′-cGAMP, 15 μg/ml). After 48 h of culture, T cells were collected, washed once with cold PBS and lysed on ice in RIPA lysis buffer (Pierce) supplemented with protease inhibitors (Roche), benzonase (Santa Cruz), and phosphatase inhibitor cocktail set V (Calbiochem). The lysates were cleared by centrifugation (15 min, 13,500 × g at 4 °C). THP-1 cells (ATCC, TIB-202), which were used as controls, were differentiated into macrophage-like cells by treatment with 150 nM phorbol 12-myristate 13-acetate (PMA; Sigma) for 24 h. Then, the PMA was removed by washing, and the cells were cultured for an additional 24 h before transfection. Next, THP-1-derived macrophages were plated at 0.3 × 106 cells/well in 24-well plates (BD Falcon) and then transfected with G3-YSD (4 µg/well, InvivoGen) complexed with Lipofectamine 2000 (Thermo Fisher Scientific) at a 1:1 ratio for 6 h before being lysed in the same manner as described for T cells. Protein concentrations were assessed by a BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific) and subsequently normalized. Proteins in the samples, along with Precision Plus Protein Dual Color Standards (Bio-Rad), were separated on 4–15% Mini-PROTEAN TGX precast gels (Bio-Rad) and transferred onto nitrocellulose membranes (Bio-Rad) by semidry transfer. The membranes were blocked in 10% sterile filtered BSA (Millipore) in TBS-T (20 mM Tris, pH 7.5; 150 mM NaCl; 0.1% Tween 20). The membranes were incubated with the relevant primary and secondary antibodies in Western BLoT Immuno Booster 1 or 2 solution (Takara Bio) and were then visualized on an Odyssey CLX-1391 imaging system (LI-COR Biosciences). The antibodies and dilutions used are listed in Supplementary Table 9.
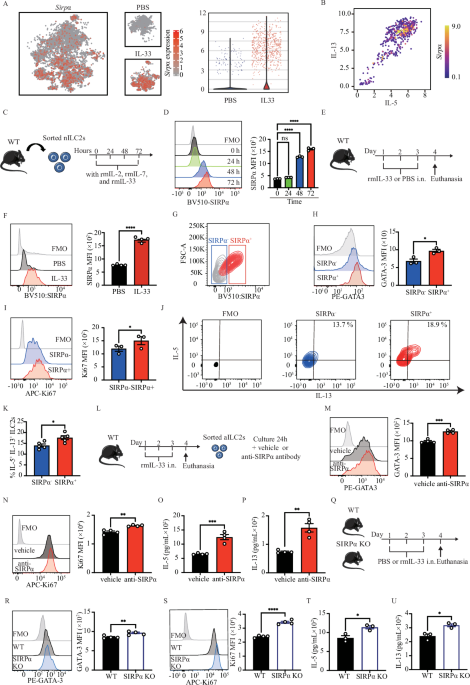
Ex vivo coculture of cDC1-CD4+ T cellsTo generate activated CD4+ T cells, naive CD4+ T cells were first activated with monoclonal antibodies against CD3 (0.1 μg/ml, clone CLB-T3/4). E, Sanquin) and CD28 (0.2 μg/ml, clone CLB-CD28/1, Sanquin) for 48–72 h. Activated and nonactivated CD4+ T cells were cultured in the presence of 10 ng/ml hIL-2, hIL-7 and hIL-15 (Miltenyi). Ex vivo-generated cDC1s were purified by flow cytometry and were then cocultured with activated CD4+ T cells for 12 h. An anti-IFNAR2 monoclonal blocking antibody (clone MMHAR-3, 5 μg/ml; PBL Assay Science), an anti-IFNGR1 blocking antibody (5 μg/ml; R&D Systems), or mouse IgG2A isotype control (5 μg/ml; InvivoGen) were added accordingly. cDC1s cultured ex vivo without CD4+ T cells were used as controls.
NanoString nCounter gene expression analysisEx vivo-cultured cDC1s were purified by flow cytometry and stimulated with universal IFNα (100 U/ml; PBL Assay Science) and IFNβ (150 pg/ml; R&D Systems), stimulated with IFNγ (10 ng/ml; InvivoGen) or left unstimulated for 12 h. An additional anti-CD40 blocking antibody (5 μg/ml; R&D Systems) was added 2 h before IFN stimulation as indicated. Then, the cells were lysed in buffer containing 1 volume of RLT buffer (QIAGEN) and 2 volumes of UltraPure™ DNase/RNase-free distilled water (Invitrogen) at a concentration of 2000 cells/μl buffer. The samples were analyzed on the NanoString nCounter® FLEX platform according to the manufacturer’s instructions. Briefly, 5 μl of lysate (10,000 cells) per condition from each donor was mixed with reporter probes, capture probes and hybridization buffer and subjected to hybridization at 65 °C for 20 h. Proteinase K (0.45 mg/ml; Thermo Fisher Scientific) was added during the hybridization step. The samples were subsequently processed on the NanoString Prep station, and the cartridges were read on the NanoString Digital Analyzer. The human host response panel (785 genes) was used. The RNA count data were normalized, scores for different pathways were calculated, and automated pathway analysis based on the expression of predefined genes was performed using nSolver software (advanced analysis module version 1.1.4). The log2-transformed output data were subsequently analyzed in R (version 4.1.2), and the ‘EnhancedVolcano’ and ‘ggplot2’ packages were used to generate the volcano plot. A heatmap of the significantly differentially expressed genes (DEGs) was generated based on the criteria of an adjusted p value of <0.05 and a log2-fold change (FC) of >1 or <−1 using Qlucore Omics Explorer (version 3.8).
siRNA transfectionNaive CD4+ T cells were first activated with monoclonal antibodies against CD3 (0.1 μg/ml; clone CLB-T3/4). E, Sanquin) and CD28 (0.2 μg/ml, clone CLB-CD28/1, Sanquin) for 36–48 h, after which the cells were washed twice with phosphate-buffered saline (PBS) before transfection. Transfection was performed in a 24-well plate. For transfection of the cells in each well, the IFNB1 siRNA complex (9 pmol, Thermo Fisher Scientific) or negative control siRNA complex (6 pmol, Thermo Fisher Scientific) was diluted in 100 μl of Opti-MEM (Gibco) supplemented with 1% FBS in the 24-well tissue culture plate (BD Falcon). Then, 1.5 μl of LipofectamineTM RNAiMAX (Thermo Fisher Scientific) was added to each well containing the diluted siRNA molecules, mixed gently and incubated for 20 min at room temperature. Next, 50,000 cells in 500 μl of complete culture medium were added to each well and incubated for 24–36 h before the gene knockdown efficiency was assessed by real-time quantitative PCR or flow cytometry.
Cas9/gRNA ribonucleoprotein (RNP) nucleofectionThis method was adapted from a previously described protocol [72]. gRNAs in the proprietary Alt-R format (Integrated DNA Technologies), i.e., a two-component gRNA composed of a crRNA (Hs.Cas9.CD40LG.1.AA and Hs.Cas9.IFNB1.1.AB) annealed to a transactivating (tracer) RNA were used. A nontargeting control gRNA in the same format was used as a control. The gRNA (100 pmol) was then incubated with recombinant Cas9 to form the Cas9 RNP complex used for T-cell transfection. Before electroporation, 150 μl of RPMI-1640 medium (Gibco) supplemented with 10% FBS and hIL-7/hIL-15 (both 10 ng/ml; Miltenyi Biotec) was dispensed into each well of a 96-well round bottom plate, and the plate was incubated at 37 °C to warm the medium. Purified naive CD4+ T cells were washed with PBS twice, counted and resuspended at 1 × 106 cells per 20 μl of P3 Primary Cell 4D-Nucleofector Buffer (Lonza) with Alt-R Cas9 Electroporation Enhancer (2 μM). Then, 5 μl of the RNP complex was added to the cell suspension and incubated for 2 min at room temperature. Next, the cells were transferred to Nucleocuvette strips (Lonza) and electroporated with pulse code FI115 in a 4D-Nucleofector Unit (Lonza). After nucleofection, the cells were immediately transferred to prewarmed medium at a density of 1 × 106 cells/well. After 24 h, the T cells were activated with monoclonal antibodies against CD3 (0.1 μg/ml; clone CLB-T3/4). E, Sanquin) and CD28 (0.2 μg/ml, clone CLB-CD28/1, Sanquin), and 48–72 h later, the gene knockout efficiency was assessed via flow cytometry.
Real-time quantitative PCRTotal RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. Five hundred nanograms of RNA was subsequently treated with ezDNase (Invitrogen) and reverse transcribed using SuperScript IV VILO Master Mix (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was diluted in nuclease-free water, and gene expression was measured with technical duplicates using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) on a QuantStudio 3 system (Thermo Fisher Scientific). The gene-specific primers used were as follows: IFNB1 forward, AGTAGGCGACACTGTTCGTG; reverse, GTCTCATTCCAGCCAGTGCT. ACTB: forward: CACTCTTCCAGCCTTCCTTC, reverse: TACAGGTCTTTGCGGATGTC.
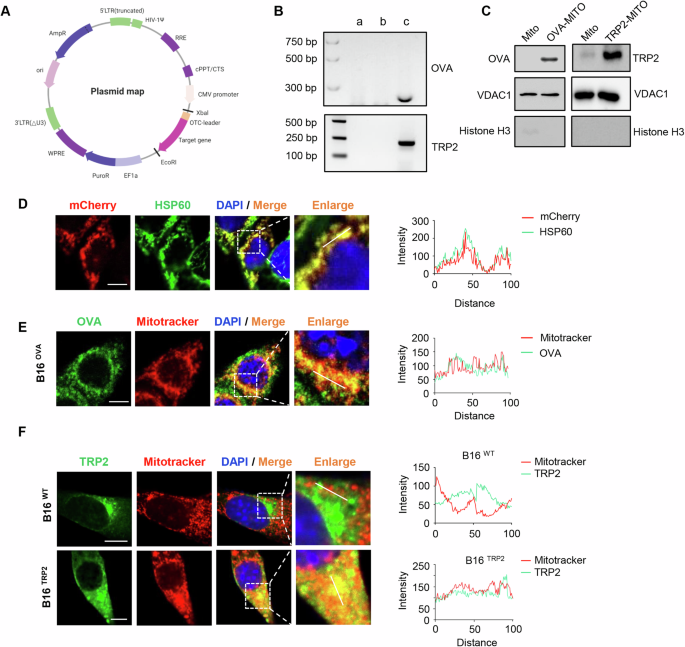
Retroviral transduction of CD8+ T cells with MART-126-35/HLA-A2-specific TCRsThis method was adapted from a previously described protocol [73]. Non-tissue culture-treated 24-well plates (BD Falcon) were coated with 10 ng/ml RetroNectin (Takara Bio) at 4 °C for 24 h, blocked with 2% bovine serum albumin (BSA; Sigma) for 30 min at room temperature (RT), and then washed with PBS twice. CD8+ T cells were cultured in RPMI-1640 medium supplemented with 10% FBS in the presence of human (h)IL-2, hIL-7 and hIL-15 (each 10 ng/ml) and human T-Activator CD3/CD28 Dynabeads (Thermo Fisher Scientific; 2 cells:1 bead) for 2–3 days before transduction. For transduction, CD8+ T cells were spun down and resuspended in retrovirus-containing medium from packaging cells supplemented with 10 ng/ml hIL-2/hIL-7/hIL-15 and plated at 0.5 × 106 cells per well. The plates were centrifuged at 800 × g for 90 min at RT in a table-top centrifuge with an acceleration setting of 3 and a deceleration setting of 0. The cells were cultured for 24 h before the virus-containing supernatant was removed and were then expanded in medium supplemented with the cytokine cocktail and CD3/CD28 Dynabeads for 7 days. Next, the Dynabeads were removed, and the cells were incubated in medium supplemented with the cytokine cocktail for 3 days before being used in CTL priming experiments.
Tumor antigen-specific CTL priming platformThis method was adapted from a protocol described in our previous publication [6]. To create conditions for antigen cross-presentation, MART-115-40 long peptide (KGHGHSYTTAEELAGIGILTV) or dead Mel526 cells were used. Mel526 cells were treated with 100 ng/ml tumor necrosis factor-related apoptosis-inducing ligand (TRAIL; Sigma) and 10 ng/ml Fas Ligand (FASL; AdipoGen) to induce apoptotic cell death. Flow sorted ex vivo HLA-A2+ cDC1s were incubated with or without activated CD4+ T cells for 2 h in IMDM supplemented with 1% FBS. Then, MART-115-40 long peptide (20 mg/ml) or dead Mel526 cells were added. After 12–16 h, the cell supernatant was removed by washing. Then, MART-126-35/HLA-A2-specific TCR-transduced CD8+ T cells were added to the culture at a ratio of 1 DC per 5-10 CD8+ T cells and cultured for 6-7 days in RPMI-1640 medium supplemented with 10% FBS and 0.2 ng/ml hIL-7/hIL-15. To trace proliferation, CD8+ T cells were labeled with CTV before being added to the CTL priming platform. Then, 50 ng/ml PMA, 1 mg/ml ionomycin (InvivoGen) and a protein transport inhibitor (BD GolgiPlug, 1:1000) were added to the culture, which was incubated for 3 h before the cells were harvested and analyzed via flow cytometry.
IncuCyte T-cell cytotoxicity assayOne day before the cytotoxicity assay, live Mel526 cells were plated in 96-well flat bottom black wall plates (Greiner) at a density of 5000 cells/well. The Mel526 cells were passed through G21 needles (BD Biosciences) before plating to prevent aggregation. The following day, the growth medium was removed from the Mel526 cells, and 100 μl of IncuCyte® Caspase-3/7 Green Apoptosis Reagent (20 μM, Sartorius) was added. Then, CD8+ T cells isolated from the tumor antigen-specific CTL priming platform via flow sorting were seeded in 100 μl of medium into the appropriate wells at effector:target ratios of 0:1, 1:1, 2:1 and 4:1. The assay plate was allowed to settle on a level surface at ambient temperature for 30 min before being placed into the IncuCyte live-cell analysis system (IncuCyte ZOOM®, Sartorius). The plates were scanned every 2 h for 18 h. Afterward, the cell suspensions were collected and analyzed by flow cytometry, and the remaining Mel526 cells were fixed with 4% PFA (Sigma), stained with crystal violet and imaged with a Zeiss Axio Imager Z1 charge-coupled device (CCD) microscope. The IncuCyte data were analyzed with IncuCyte® S3 software (version 2018B). One-way repeated measures ANOVA in SPSS (IBM) was used to determine the significance of differences. A P value of <0.05 was considered to indicate statistical significance.
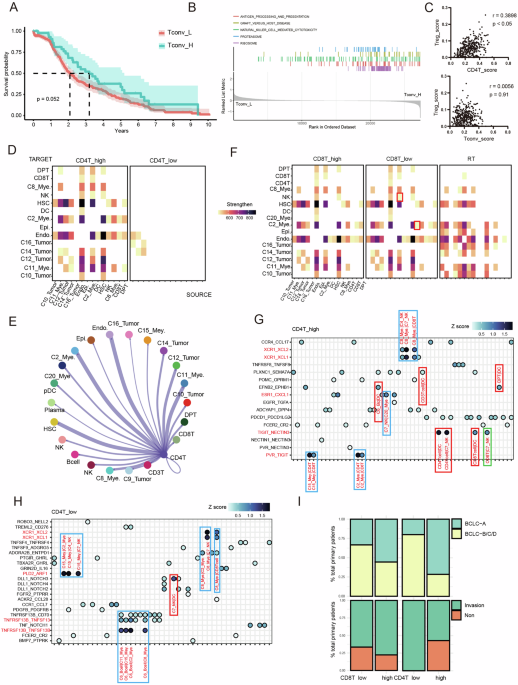
Meta-analysis of single-cell (sc) mRNA sequencing data(1) Preprocessed scRNA-seq data of immune cells from 8 primary breast tumors[37] were downloaded from the Gene Expression Omnibus (GEO). After importing the data into R (version 4.1.2) using the ‘Seurat’ package (version 4.0.0), variable features that exhibited high cell-to-cell variation in the dataset were identified using the FindVariableFeatures function. Scaling of the variable features was performed prior to dimensionality reduction using PCA. The clusters were identified using the FindCluster function in Seurat, and the nonlinear dimensionality reduction technique UMAP was used to visualize the cells in two-dimensional space. After clustering, effector-memory CD4+ T cells (CD4+ T_E(M)) and naive CD4+ T cells (CD4+ T_naive) were selected for further analysis. The FindAllMarkers function in Seurat was used to identify DEGs. The genes that were detected in 25% of the cells of in the cluster and had a log2FC of > 0.5 between two groups of cells, with an adjusted P value of < 0.05, were considered to be significant DEGs. Gene set enrichment analysis (GSEA) was performed, and a tumor-infiltrating CD4+ T-cell-enriched IFN-1 signature was identified. (2) Preprocessed scRNA-seq data from 10 studies were downloaded from GEO or from EMBL-EBI (www.ebi.ac.uk/arrayexpress). The data were imported into R (version 4.1.2) using the ‘Seurat’ package (version 4.0.0). Datasets of CD4+ T cells and CD8+ T cells from primary tumors were first selected using the Subset function based on the annotations provided by the authors of those studies and were then integrated using the Merge function. Next, the integrated data were further filtered based on cells with between 200 and 5000 transcripts and with fewer than 5% mitochondrial gene events. After removing unwanted cells from the dataset, a standard workflow for integrated data from the ‘Seurat’ package was employed. Final cluster annotation was manually performed based on the top 50 upregulated DEGs that were identified after cluster identification using the FindCluster function and differential expression analysis using the FindAllMarkers function. The nonlinear dimensionality technique UMAP was used to visualize the cells in two-dimensional space. The average expression of the IFN-I signature in tumor-infiltrating CD4+ T cells (Supplementary Table 3), the tumor-infiltrating CD4+ T-cell signature in CE9 [19] and the CXCL13+ Tht signature [40] (Supplementary Table 5) in tumor-infiltrating conventional CD4+ T cells were determined with the AverageExpression function. The ‘ggplot2’ package was subsequently used to generate a heatmap. The heatmap depicting the expression of the tumor-reactive T-cell signature [39] (Supplementary Table 5) in tumor-infiltrating CD4+ and CD8+ T cells was generated using the DoHeatmap function. The frequency of each cluster among tumor-infiltrating CD4+ or CD8+ T cells in each patient was determined in R. Spearman rank correlation coefficients were calculated using GraphPad Prism (version 9.0). (3) Trajectory inference and pseudotime calculations were performed using the ‘Monocle 3’ package [74] as part of the Seurat Wrappers workflow on the integrated scRNA-seq Seurat objects. First, the as.cell_data_set function was used to convert the Seurat object to a Monocle 3 object; next, the cluster_cells function was used to group the cells into clusters/partitions; then, the learn_graph and order_cells functions were used to model the relationships between the clusters as a trajectory of gene expression changes. The CD4+_CCR7 cluster (white circle) was used as the root_cells node (beginning of the biological process). The black circles indicate branch nodes in which cells can travel to one of several outcomes, and the end of each black line end corresponds to a different outcome (i.e., cell fate) of the trajectory. (4) Preprocessed scRNA-seq data of immune cells from HPV+ oropharyngeal squamous cell carcinoma (OPSCC) patients were acquired in our previous study [43]. Patients were stratified into the immune response-positive (IR+, n = 6) and immune response-negative (IR−, n = 4) groups based on the presence or absence, respectively, of tumor-specific T-cell infiltration. The data were imported into R (version 4.1.2) using the ‘Seurat’ package (version 4.0.0). Datasets of tumor-infiltrating FOXP3−CD4+ T cells or CD8+ T cells were first selected using the Subset function based on the annotations in our previous study. Subsequently, variable features that exhibited high cell-to-cell variation in FOXP3−CD4+ T cells or CD8+ T cells were identified using the FindVariableFeatures function. Eight subclusters were identified within FOXP3-CD4+ T cells, and seven subclusters were identified within CD8+ T cells using the FindCluster function in Seurat, and the nonlinear dimensionality technique UMAP was used to visualize the cells. The average expression of the tumor-infiltrating CD4+_CXCL13 and CD4+_MKI67 signatures that we identified (Supplementary Table 4), as well as the tumor-infiltrating CD4+ T-cell signature in CE9 [19] and the CXCL13+ Tht signature [40] (Supplementary Table 5), in the FOXP3−CD4+ T-cell subclusters were obtained using the AverageExpression function. A heatmap depicting the expression of the IFN-I signature in tumor-infiltrating CD4+ T cells (Supplementary Table 3) in clusters 3 and 4 of FOXP3-CD4+ T cells and a heatmap depicting the expression of the tumor-reactive T-cell signature [39] (Supplementary Table 5) in CD8+ T cells were generated using the DoHeatmap function. The cell numbers per cluster in each patient were determined in R. The proportion of each cluster in each patient was then calculated, and statistical analysis was performed using GraphPad Prism (version 9.0).
Gene set enrichment analysis (GSEA)The Log2FC values of the 577 DEGs between cDC1s cultured with activated CD4+ T cells and cDC1s cultured with naive CD4+ T cells identified by our scRNA-seq analysis or the log2FC values of the 542 DEGs between tumor-infiltrating CD4+ effector-memory T cells and naive CD4+ T cells identified in the scRNA-seq data of 8 primary breast tumors [37] were used for GSEA. GSEA software (version 4.2.2) (http://broadinstitute.org/gsea) and the Reactome pathway database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) were used with default parameters to calculate enrichment and generate GSEA plots.
Spatial transcriptome analysisPreprocessed spatial transcriptomics datasets of breast carcinoma specimens were downloaded from 10x Genomics (https://www.10xgenomics.com/spatial-transcriptomics/). After the data files were uploaded into Cell Loupe browser software 5.0, the Gene/Feature Expression mode was chosen, and the data were scaled according to the average log2Feature values. The IFN-I signature from tumor-infiltrating CD4+ T cells (Supplementary Table 3), the tumor-infiltrating CD4+ T_CCR7, CD4+ T_LMNA, CD4+ T_ISG15, CD4+ T_CXCL13 and CD4+ T_MKI67 signatures (Supplementary Table 4), the CXCL13+ Tht signature [40] (Supplementary Table 5), the tumor-infiltrating CD4+ T-cell signature in CE9 [19] (Supplementary Table 5), and the cDC1 “help” signature [6] were uploaded, and the distributions of the different signatures were visualized.
Survival analysis in The Cancer Genome Atlas (TCGA) datasetsSurvival analysis was carried out using the top 20 upregulated DEGs in the CD4+ T_CCR7, CD4+ T_LMNA, CD4+ T_ISG15, CD4+ T_CXCL13 and CD4+ T_MKI67 clusters (Supplementary Table 4). We assessed patient OS times and tumor gene expression profiles for combined datasets of BRCA, COAD, LIHC, LUAD, OV, READ and SKCM patients (n = 3156) in the TCGA database using the GEPIA2 computational workflow [75] based on the UCSC Xena platform (http://xena.ucsc.edu). Briefly, OS analysis was based on the log-rank hypothesis test (the Mantel–Cox statistical test), with estimation of Cox proportional hazard ratios (HRs) and 95% confidence intervals, accompanied by a Kaplan‒Meier (KM) plot. Herein, a threshold of the quartile signature expression level was used to divide the patients into the high-expression and low-expression subcohorts.
Bulk mRNA analysisThe preprocessed RNA-seq TPM matrix was downloaded from the supplementary information of Liu et al. (https://www.nature.com/articles/s41591-019-0654-5#Sec31) and imported into R (version 4.1.2). Genes with a TPM of 0 in more than 25% of the samples were removed. After filtration, the ‘GSVA’ package [76] (version 4.2) was used to perform GSEA with the defined gene sets/signatures (Supplementary Table 4) in responders and nonresponders. Responders were defined as patients who achieved complete response (CR) or partial response (PR). The enrichment score for each patient (n = 120) was then used to calculate p values using the Wilcoxon signed-rank test. A P value of <0.05 was considered to indicate statistical significance.
Statistical analysisThe data, excluding mRNA sequencing data obtained from public datasets and IncuCyte cytotoxicity assays, were analyzed using GraphPad Prism (version 9.0), and the Mann‒Whitney U-test or one-way ANOVA was used to determine the significance of differences among samples or groups. The data are presented as the means ±SEMs. A P value < 0.05 was considered to indicate statistical significance.
Materials and data availabilityThis study did not generate new unique reagents. This paper analyzed existing, publicly available data. The accession numbers of the datasets are listed in the key resources table. The TCGA cohorts used in the survival analysis can be accessed via http://gepia2.cancer-pku.cn/#index. The Reactome database can be accessed via https://reactome.org/. All packages used to analyze sequencing data are publicly available and are listed in Supplementary Table 9. All the other primary data and materials that support the findings of this study are available from the corresponding author upon request.
留言 (0)