Continuous cell lines
VERO C1008 (Vero E6) cells (ATCC CRL-1586™) were maintained in Dulbecco’s Modified Eagle Medium, high glucose, pyruvate, L-glutamine (Gibco) supplemented with 10% foetal bovine serum (CellSera), 10 mM HEPES (MP Biomedicals), and 1 × penicillin–streptomycin (Gibco) at 37 °C and 5% CO2.
BHK-21 cells (ATCC CCL-10™) were maintained in Minimum Essential Media supplemented with 10% foetal bovine serum, 10% tryptose phosphate broth, 10 mM HEPES, 2 mM glutamine (Gibco), and 1 × penicillin–streptomycin at 37 °C and 5% CO2.
Stem cell–derived human neural cultures
The HDF51i-509iPSCs were differentiated into NPCs as described previously (Murphy et al. 2017). NPCs were then maintained in STEMdiff™ Neural Progenitor Medium (STEMCELL Technologies) supplemented with 1 × STEMdiff neural progenitor supplement A (STEMCELL Technologies) and 1 × STEMdiff neural progenitor supplement B (STEMCELL Technologies) at 37 °C and 5% CO2. Neural differentiation was initiated by replacing 50% v/v of the culture medium every 2–3 days with BrainPhysTM neural medium (STEMCELL Technologies) supplemented with 2% v/v Neurocult™ SM1 Neuronal Supplement (STEMCELL Technologies), 1% v/v N2 Supplement-A (STEMCELL Technologies), 20 ng/mL human recombinant brain-derived neurotrophic factor, 20 ng/mL human recombinant glial cell line–derived neurotrophic factor (Peprotech), 1 mM dibutyryl cyclic-AMP sodium salt (Sigma-Aldrich), and 200 nM ascorbic acid (STEMCELL Technologies), making complete BrainPhys™ neural medium (Sundaramoorthy et al. 2020a).
Viruses
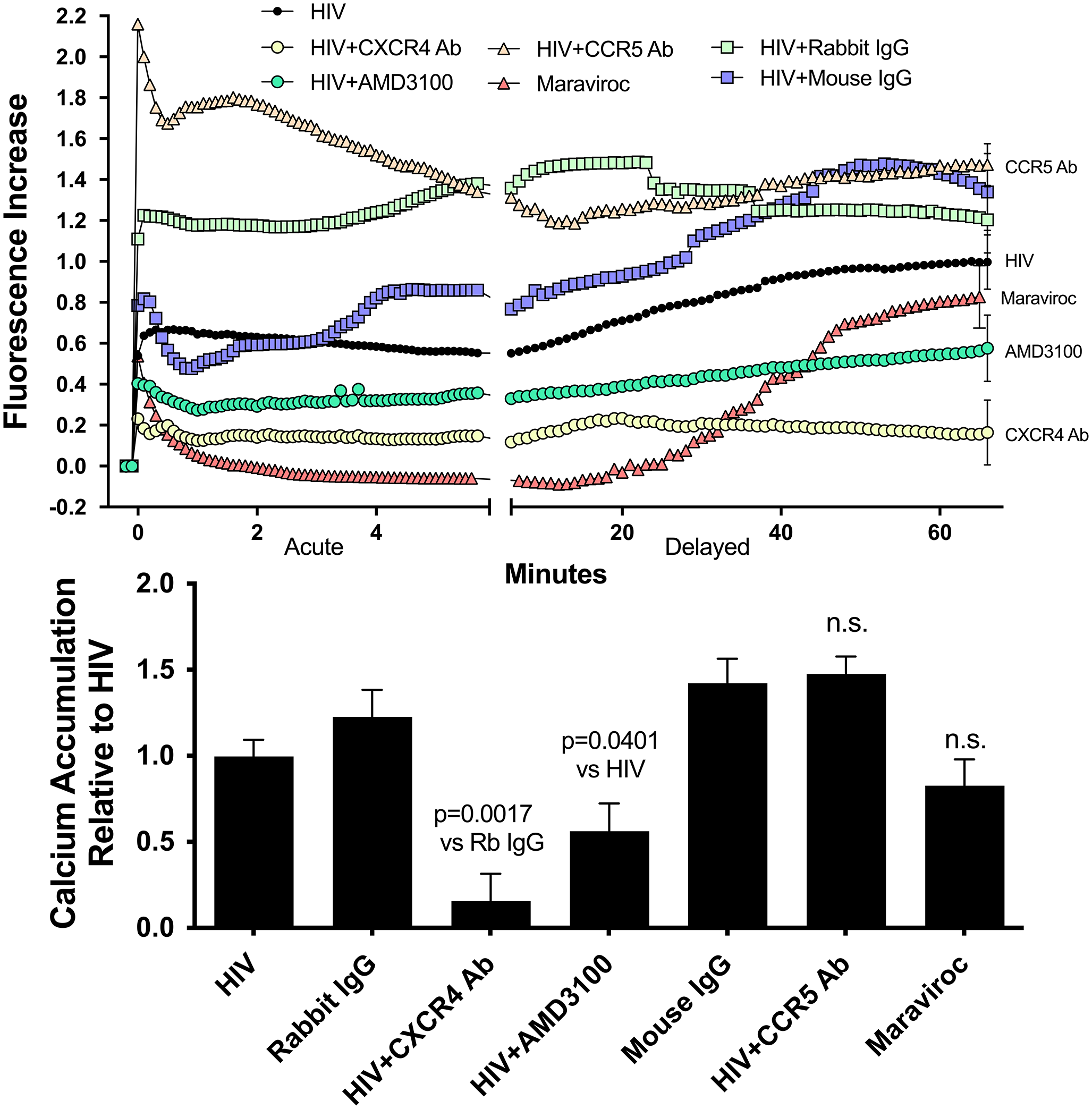
Severe acute respiratory syndrome coronavirus, HKU-39849 isolate (SARS-HK) (NCBI accession: AY278491.2), and SARS-CoV-2 SARS-CoV-2/human/AUS/VIC01/2020 (VIC01) (NCBI accession: MT007544.1), SARS-CoV-2/human/AUS/VIC02/2020 (VIC02) (NCBI accession: MT450919.1), and B.1.617.2 variant SARS-CoV-2/human/AUS/18440/2021 (Delta) strains were propagated in VERO C1008 cells, harvested by freeze–thaw ≤ 3 days post-inoculation, clarified by centrifugation, and stored at − 80 °C. Sarbecovirus titres were quantitated by titration in VERO C1008 cells. Infectious tissue culture supernatants were titrated tenfold and added to Vero C1008 monolayers in quadruplicate. Plates were incubated at 37 °C and 5% CO2 for 5 days, and cytopathic effect was visualised by light microscopy.
Rabies virus (RABV) CVS-11 strain was propagated in BHK-21 cells, harvested by freeze–thaw 5 days post-inoculation, clarified by centrifugation, and stored at − 80 °C. Virus titre was quantitated by titration in BHK-21 cells. Briefly, infectious tissue culture supernatants were titrated tenfold and added to BHK-21 monolayers in quadruplicate and incubated for 5 days at 37 °C and 5% CO2. RABV-infected monolayers were fixed with 4% paraformaldehyde (Sigma) in phosphate-buffered saline (PBS) (Thermo Fisher Scientific) at room temperature for ≥ 1 h or 4 °C overnight. Fixed monolayers were washed twice with PBS and FITC-conjugated anti-RABV monoclonal antibody cocktail (1:200) (Fujirebio Diagnostics) in PBS supplemented with 0.005% Evan’s Blue (Sigma), and 1% bovine serum albumin was added, and monolayers were incubated at 37 °C for 30 min. Monolayers were washed three times with PBS before adding PBS to each well. Fluorescence was visualised by fluorescent microscopy.
Virus stock titres were calculated by Reed and Muench method (Reed and Muench 1938). All work with infectious sarbecoviruses was performed at PC4/BSL4. All work with inactivated sarbecoviruses and RABV was performed at PC3/BSL3.
Sarbecovirus infection of human iPSC–derived NPCs and neural cultures
HDF51i-509 NPCs (passage 11) were seeded at a density of 8 × 104 cells/well in complete Neural Progenitor Medium in 24-well plates coated with 15 µg/mL poly-L-ornithine and 10 µg/mL laminin with or without glass coverslips (13 mm; Menzel Gläser). Undifferentiated NPCs were infected 24 h after seeding, or NPCs were differentiated for up to 50 days before infection. Neural cultures were infected at a multiplicity of infection (MOI) of 1 for up to 72 h. Briefly, appropriate volume of viral inoculum to infect at MOI 1 for each well was prepared in a total of 150 µL of BrainPhys™ neural medium. After removing the existing culture media in each well, 150 µL of diluted viral inoculum was added to the well, incubated at 37 °C and 5% CO2 for 30 min, and then, additional 500 µL of BrainPhys™ neural medium was added. Infected cultures were incubated at 37 °C and 5% CO2 for up to 72 h.
Ex vivo model of interconnected human neurons using microfluidics
Xona microfluidic devices (Xona Microfluidics, Cat#SND450) were sterilised and plasma bonded to glass coverslips (24 × 40 mm; Menzel Glaser) using a plasma cleaner (PDC-32G-2, Harrick Plasma). After bonding, the devices were coated with 15 µg/mL poly-L-ornithine (Sigma) and 10 µg/mL laminin (Sigma). HDF51i-509 NPCs were seeded in both panels of a microfluidic device at a density of 8 × 104 cells per panel in BrainPhys™ complete neural differentiation media to initiate differentiation. The media in the wells were replenished every 2–3 days and the differentiation was continued for up to 21 days.
Microfluidic chambers were infected as described previously (Sundaramoorthy et al. 2020a) with sarbecoviruses or RABV at a MOI 1 at day 20–21 of differentiation. Briefly, media from the panel to be infected was removed and the appropriate volume of viral inoculum in BrainPhys™ neural medium required to infect at MOI 1 was added. A unidirectional flow of media was strictly maintained by a higher volume of media in the non-infected panel (200 µL) and a lower volume in the infected panel (100 µL). Sarbecovirus- or RABV-infected microfluidic chambers were incubated at 37 °C and 5% CO2 for 24 h.
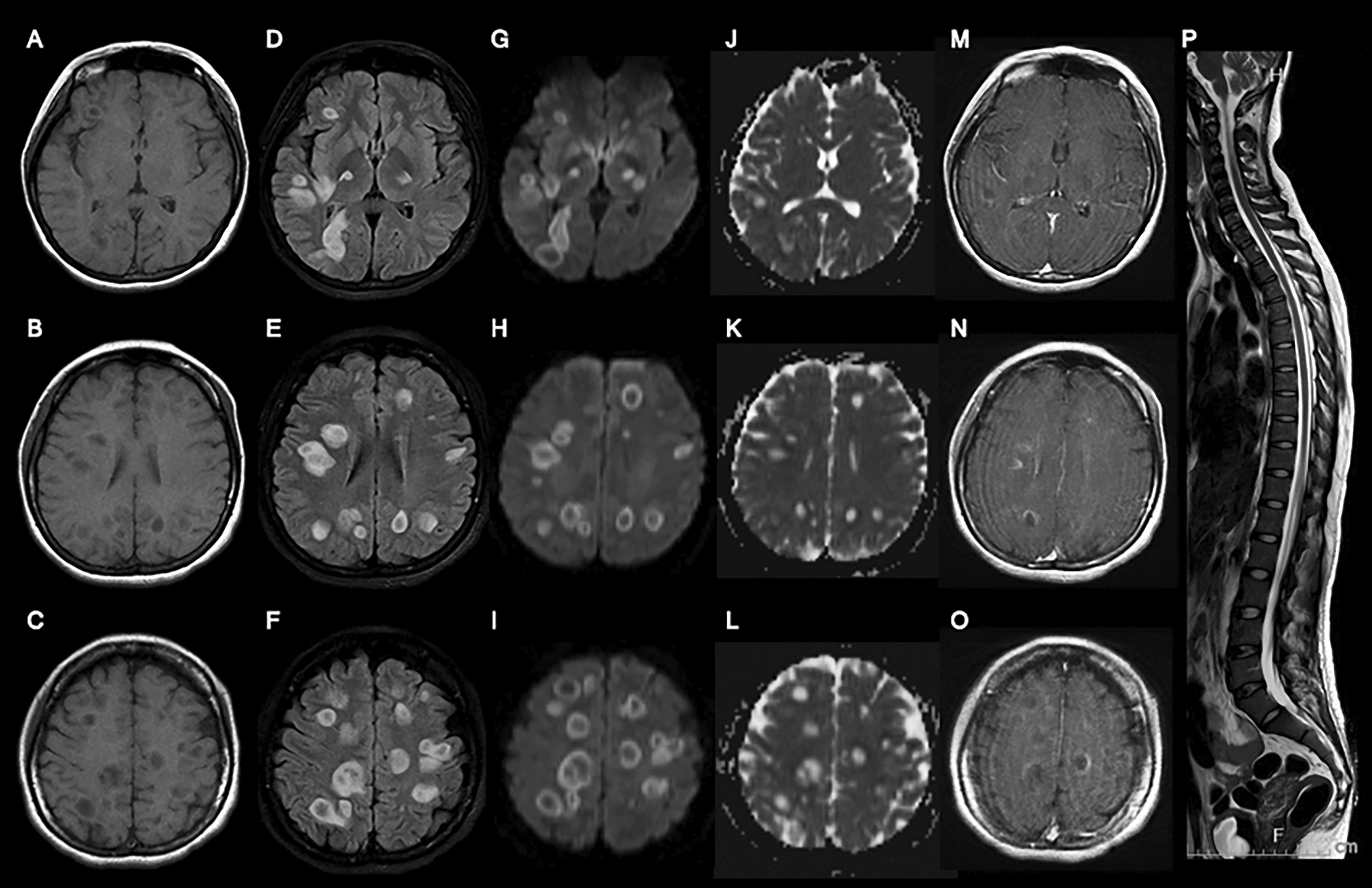
Replication of SARS-CoV-2 in ferret nasal neuroepithelium
SARS-CoV-2 VIC01 infection of ferrets is described in Au et al. (2022). Briefly, ferret turbinates at 7 days post infection were fixed in 10% neutral buffered formalin (Australian Biostain). Preserved ferret turbinates were processed according to routine histological methods, embedded in paraffin wax (Leica Biosystems), and 4-µm serial sections were mounted on glass slides (Menzel Gläser). Tissue sections were deparaffinised using routine histological methods. Antigen retrieval was performed using Target Retrieval Solution, high pH (Dako) at 97°C for 30 min on a PT Link pre-treatment module (Agilent).
Immunofluorescence staining and confocal imaging
Sarbecovirus-infected HDF51i-509 iPSC-derived neural cultures were fixed with 4% paraformaldehyde in PBS at room temperature for ≥ 1 h or overnight at 4 °C and labelled using immunocytochemistry. Fixed cells were washed twice with PBS, permeabilised with 0.1% Triton X-100 (Sigma) in PBS for 10 min, washed with PBS, and blocked with 0.5% bovine serum albumin (Sigma Bovogen Biologicals) in PBS for 30 min. Primary antibody incubation was performed overnight at 4 °C in block buffer, washed three times with PBS, and secondary antibody incubation was performed for 1 h at room temperature in block buffer. The following primary antibodies were used: 1:2000 hyperimmune horse anti-SARS polyclonal antiserum (Yu et al. 2008), 1:500 rabbit anti-SARS-CoV-2 Spike S1 (Sino Biological, cat#40,150-R007), and 1:1000 mouse anti-beta III tubulin (TuJ1), clone 2G10 (Abcam). The following secondary antibodies were used: 1:500 goat anti-horse IgG (H&L)-FITC (Abcam), 1:200 goat anti-rabbit IgG (H&L)-Alexa Fluor (AF) 568 (Invitrogen), and 1:200 goat anti-mouse IgG (H&L)-AF 647 (Invitrogen). Following secondary antibody incubation, monolayers were washed twice with PBS, then twice with deionised water before 1:4000 DAPI (Invitrogen) was added for 10 min. Monolayers were washed twice with deionised water and mounted on glass slides (Menzel-Gläser) using Vectashield (Vector Laboratories).
RABV-infected HDF51i-509 iPSC-derived neural cultures were fixed and labelled using immunocytochemistry as above with the following modification: 1:3000 rabbit anti-RABV nucleoprotein monoclonal antibody (Rahmadane et al. 2017), 1:1000 chicken anti-MAP2 (Abcam) monoclonal antibody, and 1:200 goat anti-chicken IgG (H&L)-AF 488 (Thermo Fisher) secondary antibody were used.
To assess sarbecovirus replication in ferret neuroepithelium, tissue sections were washed twice with PBS before permeabilising with 0.1% Triton X-100 in PBS for 10 min. Tissue sections were blocked with 5% bovine serum albumin and 0.1% Triton X-100 in PBS overnight at 4 °C. Primary monoclonal antibody incubation was performed overnight at 4 °C in block buffer in a humified chamber, washed three times with PBS, and secondary antibody incubation was performed for 3 h at room temperature in block buffer. The following primary antibodies were used: 1:10,000 rabbit anti-SARS-CoV-2 nucleocapsid (Sino Bio), 1:1000 mouse anti-SARS-CoV-2 nucleocapsid (Sino Bio), 1:1000 mouse anti-TuJ1, and 1:3000 rabbit anti-olfactory marker protein (OMP) [EPR19190] (Abcam). The following secondary antibodies were used: 1:200 goat anti-rabbit IgG (H&L)-Alexa Fluor (AF) 488 and 1:200 goat anti-mouse IgG (H&L)-AF 568 (Invitrogen). Following secondary antibody incubation, tissue sections were washed twice with PBS, then twice with deionised water before 1:4000 DAPI (Invitrogen) was added for 10 min. Monolayers were washed twice with deionised water and mounted on glass slides using Vectashield.
Confocal imaging was performed using a LSM 800 inverted confocal microscope (ZEISS). Images were taken as Z-stacks with or without tile scan and then maximum intensity projection was generated. Airyscan™ module was used to image ferret nasal turbinate tissues using 63 × objective with Z-stacks. All the confocal imaging and processing were performed using ZEN 2.5 Blue software (ZEISS, Oberkochen, BW, Germany).
Quantification of infected neurons from confocal images
Confocal images of neural cultures stained with TUJ1 (neuron, red), viral antigen (green, anti-SARS2-S1; magenta, anti-SARS), and DAPI (blue, nucleus) were taken with 20 × objective covering 100–400 neural cells in a single image. Total number of cells (DAPI stained nuclei) were counted using ImageJ particle analyser plugin and watershed function, with the following parameters: size (inch2) 0.005–infinity and circularity 0.00–1.00. The number of TUJ1 and viral antigen–positive neurons were counted manually in the same image to determine the % of infected neurons.
Cell-associated sarbecovirus RNA quantitation
RNA was extracted from sarbecovirus-infected neural cultures using MagMAX Viral RNA Isolation Kit (Applied Biosystems) using a KingFisher magnetic particle processor (Thermo Fisher Scientific). cDNA was synthesised using SuperScript IV VILO Master Mix with ezDNase enzyme (Thermo Fisher Scientific) according to manufacturers’ instructions.
Cell-associated sarbecovirus RNA quantitation was performed in duplicate by quantitative real-time PCR using AgPath-ID One-Step RT-PCR kit (Applied Biosystems), sarbeco E gene reverse primer and sarbeco hydrolysis probe (Corman et al. 2020) with the 3′ quencher modified to MGB, and a modified E gene forward primer (Marsh et al. 2021) on a QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific) according to manufacturers’ instructions. Sarbecovirus E gene copy numbers were calculated using cycle threshold (CT) and a standard curve of known copy number.
Statistical analysis
Statistical analyses (ordinary one-way ANOVA with Tukey’s multiple comparison) were performed using GraphPad Prism, version 9.3.1 (GraphPad Software). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001.


留言 (0)