
記住我



We will conduct an assessor blind randomized, controlled, two-arm clinical trial to assess whether the combination of a low-calorie diet and PFMT will optimize the results obtained with diet therapy as a single intervention to decrease UI prevalence reports and severity in obese women (primary outcome). The secondary outcomes of this study are the impact of the protocol on PFM function, women’s self-perception of their PFM, adherence, and satisfaction with the treatment. This protocol will be reported according to the Protocol Items Standard: Recommendations for Intervention Trials checklist (SPIRIT) 2013: items recommended for a clinical trial protocol and related documents. It is a superiority trial.
EthicThe study was approved by the Human Research Ethics Committee of the University of São Paulo—Hospital das Clínicas of the Ribeirão Preto Medical School (HCFMRP / USP) No. 16379919.5.0000.5440 on July 9, 2019, and registered in the Clinical Trials—NCT04159467.
Any major deviation or modification of the original protocol will be reported in the Clinical Trials website and the trial records will be continuously updated as necessary.
The risks of this research basically include the possible embarrassment of the participant in answering questions related to symptoms of urinary incontinence, intestinal and vaginal pain, and embarrassment during the physical examination (vaginal palpation), in addition to feeling some discomfort during the examination and of the possible breach of confidentiality during the research.
The benefits of this research are that the participant will receive information about the anatomy of the pelvic floor, its functions, and dysfunctions, she will learn to contract this musculature and know if she is doing it correctly, contributing to prevent and/or treat its dysfunctions, in addition to also receiving a program of training for 12 weeks (if selected for group II) to strengthen the pelvic floor muscles. If you are selected for group I, you will be invited after 12 weeks to receive the same treatment as in group II.
For ethical reasons, we did not plan to prohibit any concomitant treatment. We included in the methods section our plan to monitor any concomitant treatment for pelvic floor disorders including urinary incontinence, but we expect to have homogeneous groups in relation to these variables provided by an adequate randomization and sample size.
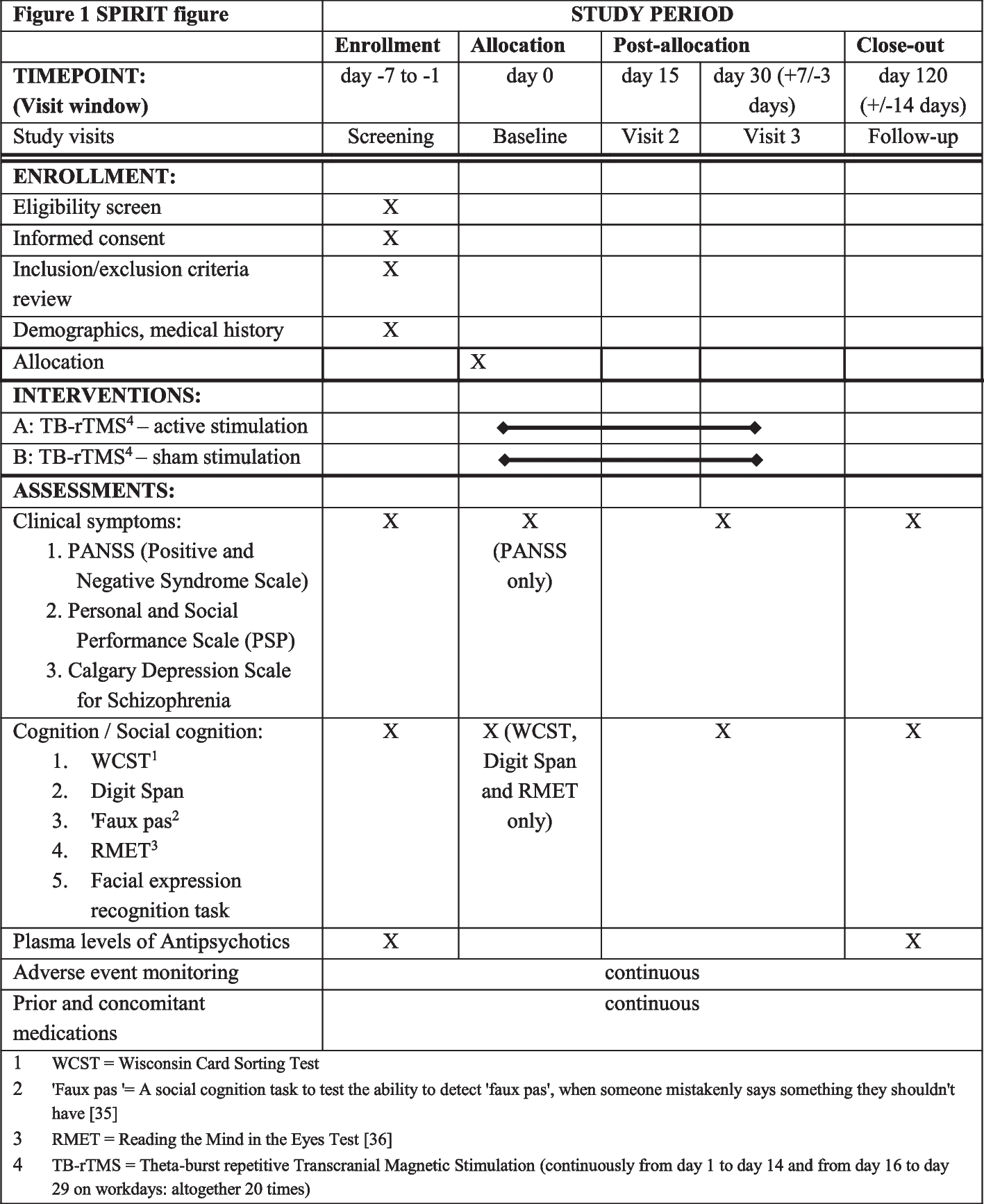
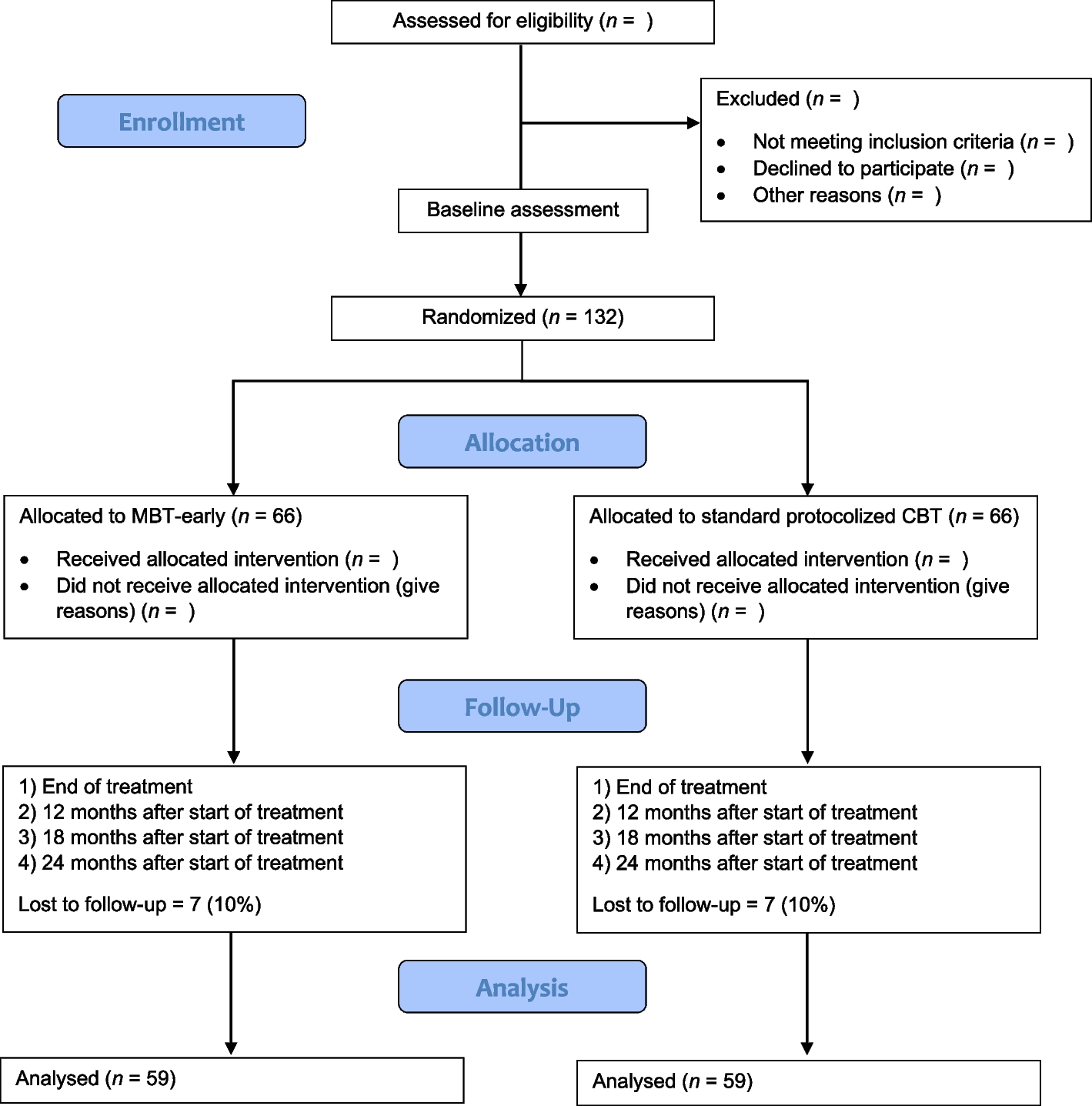
Figure 1 shows the CONSORT (Consolidated Standards of Reporting Trials) flowchart, enrollment schedule, interventions, and study evaluations. Figure 2 shows the checklist using the SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) used in the study.
Fig. 1
Flowchart for selecting women for the randomized clinical trial, following the CONSORT
Fig. 2
Study evaluation schedule (SPIRIT). Source: According to the SPIRIT 2013 statement: Standard Protocol Item Definition for Clinical Trials
ParticipantsObese women with UI who are about to start a low-calorie diet program for weight loss will be recruited in the bariatric surgery sector at the Clinics Hospital of Ribeirão Preto Medical School (HCFMRP-USP).
Inclusion criteriaWomen over 18 years, with a body mass index (BMI) > 30 kg/m2; reporting stress urinary incontinence, urgent urinary incontinence, or mixed urinary incontinence in the past 4 weeks assessed by the ICIQ-SF; who are able to contract the pelvic floor muscles (assessed using vaginal palpation with a PFM contraction ≥ 2 according to the modified Oxford grading scale); and without any cognitive impairment.
Exclusion criteriaThe participants will be excluded of the study if they withdraw their consent to participate in the study or if they become pregnant.
Criteria of discontinuingThe criteria of discontinuing the trial for a given participant include withdrawal of consent to participate and becoming pregnant during the study.
Sample size calculationThe sample calculation was estimated considering a significance level of 5%, power of 20%, and difference of 4 points on the final score of the primary outcome, ICIQ-SF, between groups at the end of treatment [10]. The value of 4 points is equivalent to the minimum value for change in the questionnaire score. Thus, the sample will be composed of 22 participants.
Statistical analysisData will be de-identified, encoded, and stored in the database using the Excel Program. The software SPSS 22.0 will be used for the analysis of data. The description of quantitative variables will be expressed in mean, standard deviation, minimum value, median and maximum value. Qualitative variables will be described in absolute and relative values (%). A 95% confidence interval and 0.05 significance will be considered for all tests. Both per-protocol and intention-to-treat analyses will be performed.
A linear mixed-effect model will be used to analyze the ICIQ-SF score. In the reassessment, individuals’ values will be considered random effects, and the groups, the times, and the interaction between them will be regarded as fixed effects. In addition, the McNemar test will verify changes in categories variables before and after the intervention. Both per-protocol analysis [11,12,13] and an intention-to-treat analysis will be also conducted.
Randomization and blinding processAn assistant researcher specially trained will be responsible only for recruiting the participants to enter the study and will give them all the information about the study, obtaining their written consent in case they agree to participate.
An assistant researcher not involved with the recruitment, intervention and assessment will generate the allocation sequence. The participants will be allocated in a group that will receive a low-calorie diet for weight loss (group I) or in another group that will receive diet + PFMT (group II). Randomization will be carried out using a computational list of random numbers and will be concealed (by means of a sealed brown envelope). Women’s data will be kept only with the assistant researcher not involved with other parts of the research process to process to protect confidentiality before, during, and after the intervention. The trial is an assessor blinded in relation to the pelvic floor muscle assessment.
The senior researcher who is not involved with the recruitment, intervention delivery, and assessment will have access to the trial data. We have planned to submit the manuscript to an open-access journal that makes all data available to readers as an appendix or under request.
Figure 1 shows the flowchart for selecting women for the randomized clinical trial, following the CONSORT.
InterventionWomen randomized to Group 1 will receive counseling from the hospital staff. They will attend 3 meetings (once a month), where a multi-professional team from the bariatric surgery outpatient clinic will address different topics related to nutrition including advice on the quality of food, mindful eating, identification of signs of hunger and satiety, and emotional triggers that lead to emotional eating. In addition, each participant will undergo individual nutritional assistance to assess food consumption, anthropometric measures, and body composition and will receive more specific guidelines on nutrition and an individualized low-calorie diet and monitoring. Outpatient care will last 12 weeks and women in this group will not receive instructions and a supervised PFMT protocol, however for ethical reasons, after the data collection is finished women belonging to this group will be invited to receive six supervised sessions of PFMT and will receive the same booklet of exercises on PFMT delivered to group II.
The participants randomized to group II will also receive the same counseling related to nutrition and also an individualized low-calorie diet given by the hospital staff; additionally, they will participate in six face-to-face physiotherapy sessions supervised by a physiotherapist in groups of a maximum of 10 participants, for 12 weeks in interspersed weeks.
In the weeks when there is no face-to-face supervised session, the same physiotherapist will encourage home PFMT by phone, calling each participant of Group II only. An intensive PFMT will be encouraged by the physiotherapist. The PFMT protocol will consist of 4 sets of ten maximum perceived voluntary contractions sustained for 6 s with a 6-s resting period between the contractions. At the end of a series of 10 contractions, five rapid contractions of the PFM will be performed. Two minutes interval will be given between each set performed.
The sets will be performed only in two positions at the face-to-face supervised sessions at the hospital: sitting and standing (2 sitting sets + 2 standing sets). The participants of this group will be instructed to perform PFMT at home at least three times a week, except on the days of supervised training. They will be instructed to follow the booklet given to them in the first meeting, in addition to the fortnightly adherence diary to the exercises to be filled weekly. Participants will also be instructed on how to perform the “the knack” maneuver, which consists of repeatedly contracting their PFM before any increase in intra-abdominal pressure during daily activities [14]. Women will also be instructed to repeatedly contract their PFM when they feel urgency. Women will fill a diary about their adherence to PFMT, and there will be a place where they are asked about any side effects or bother related to the intervention. At the last assessment, they are asked about again about any unintended effects related to the trial intervention. The type and number of unintended effects will be fully reported as an important result of the trial. Although adverse effects of these type of intervention is not very common or severe, all the participant is assured they can discontinue their participation at any time for any reason, including any adverse effects (Table 1). Table 1 presents detailed information about the protocol of intervention.
Table 1 Stages of the study and information about the 12 weeks training AssessmentPrimary outcome measuresSelf-report of urinary incontinence will be measured by question 3 of the ICIQ-SF. Women will be considered incontinent if they choose options 1, 2, 3, 4, or 5 of question 3. Women will be considered continent if they choose option 0 in question 3.
The severity and impact of urinary incontinence on women’s quality of life will be measured by the ICIQ-SF score.
Other outcome measuresThe adherence to home PFMT sessions will be monitored by the physiotherapist from a diary where women will register their adherence to home training. This diary will be attached to the leaflet that will be given to women in the first session. The diary will be collected by the physiotherapist every fortnight.
The PFM function will be assessed using vaginal palpation by a trained physiotherapist who will not be involved with the intervention and will not be aware of women’s allocation in the groups. Before the start of the palpation exam, the woman will receive detailed information about the anatomy, function, and dysfunction of the PFM and how to contract them correctly. The evaluation will be carried out with the participant in the supine position with the hip and knee semi-flexed. The participant will be asked to pull their PFM in and up as hard as possible, and then she will be instructed to relax them completely. Contraction of PFM will be classified as absent or present. The ability to contract the PFM will be considered present when there is occlusion and/or occlusion and elevation of the PFM towards the pubic symphysis and absent when there is no perceived internal movement. Additionally, muscle contraction will be graded according to the Modified Oxford Grading Scale (MOGS) [15, 16]. Contractions of muscles such as gluteus, adductor, and abdominal will be discouraged by the examiner.
Self-perception of women’s PFM contraction will be assessed at the time of the physical evaluation when the examiner will ask the participant if she feels she is able to contract her PFM and she will be asked to estimate the intensity of her PFM according to the MOGS that will be presented to her [17].
Participants’ subjective satisfaction with the treatment will be measured by the visual analog scale (VAS), which is an estimate on a numerical scale where 0 represents no satisfaction at all and 10 means the maximum possible satisfaction with the treatment.
In relation to satisfaction with the interventions, participants will be asked to answer the following questions: “Are you satisfied with the treatment you received for UI?”, “Would you do this treatment again?”, “Would you recommend this treatment?”, “Would you pursue another treatment?”. The options of answers to these questions are yes or no.
In order to identify barriers to treatment, the participants will be asked to answer the following questions: “Do you identify any barriers or difficulties in adhering to the treatment you received for urinary incontinence? If any please list the barriers.”
Descriptive and control variablesThe descriptive and control variables were divided into clinical variables (diabetes, hypertension, constipation, number of pregnancies, deliveries, cesarean sections, vaginal delivery; use of contraceptives and previous pelvic surgeries) sociodemographic variables (age, education, marital status, economic, smoking occupation, use of alcoholic beverages, physical exercise and use of caffeine) and anthropometric measures (height and body mass). The descriptive and control variables will be acquired from women’s self-report. The study variables are shown in Fig. 2 and Table 2.
Table 2 Outcome measures and time points of the study assessmentData collectionThe data will be collected in an interview format using a self-reported validated questionnaire and PFM exam. A trained assistant researcher will conduct an interview and a PFM assessment. All assessments will be performed (at baseline) and after 12 weeks for both groups and will be performed by the same researcher that will be blind about women’s group allocation.
留言 (0)