
記住我

This study is designed as an experimental single-centred, two-arm parallel group randomised controlled trial over 11 months with a 1:1 allocation ratio.
Study settingThe study will be conducted at a regional hospital in a rural part of Northern Norway. Participants will be recruited from 3 surgical departments: the orthopaedic department, the urology and vascular/thoracic surgery department and the gastro, gynaecological, breast and surgical endocrine department. The hospital serves a broad range of mental and somatic healthcare and substance abuse patients from 20 municipalities in the region. The annual budget of the hospital is approximately 5 billion Norwegian kroner. The number of employees is about 4800. Due to logistical reasons affecting the project, which will be carried out in a rural area, patients eligible for inclusion in the study must live in 1 of 9 predetermined municipalities closest to the hospital and the location of the research team. The 9 chosen municipalities have between 1000 and 53,000 inhabitants. The Norwegian healthcare system (both primary and secondary) is publicly funded and ensures all residents with free access to care.
Participants and recruitment strategySurgical patients over 65 years of age will be eligible to participate in the study if they meet the following criteria:
1)They are inpatients.
2)Their home address is one of the municipalities affiliated with the selected hospital.
3)They live at home and are due to return home after discharge directly from hospital or after a short-term training stay at a training centre before returning home.
4)They can read and understand Norwegian.
5)They have consent competence.
6)They have a mobile phone or PC/tablet able to connect to the Internet.
7)They have a body mass index < 24.
Following guidelines by Helsedirektoratet [54] and using NRS 2002 [55] for nutrient screening, the study will be based on the premise that all bedridden older adults undergo nutrition screening during their hospital stay and therefore have a known BMI. Patients receiving only liquid diets, such as tube feeding or intravenous nutritional support, and terminally ill patients will be excluded from the study.
The first author will have access to the department’s record system and will be responsible for informing and obtaining consent from eligible patients. The first author will enter the data into a spreadsheet and assign the participants to the intervention or control groups after the randomisation has been done. Furthermore, a trained external data collector who is not involved in the project will perform all the measurements.
Sample sizeWe base our calculation of the sample size on the effect estimates of the primary outcome of the combined physical scale from the feasibility study, as estimates were not available in the literature [53]. Alpha was set at 0.05, and the beta was set at 0.20. The estimated effect size was calculated to be 7 points on a scale ranging from 0 to 100, and the standard deviation of the population outcome was 18. The patients will be distributed with 50% in the control and intervention groups. Using the sample size-means calculator [56], the necessary sample size was calculated to be 104 in each of the 2 groups. Based on the feasibility study [53], we estimated a drop-out rate of 32%. Therefore, we need to recruit 138 participants for each group.
RandomisationComplete randomisation will be achieved using the free software program RANDOM.ORG [57]. The first author will use the program to allocate participants to the inclusion group or the control group randomly. The study will be blinded to the care providers and data collectors, including the external data collector who will perform the outcome assessors and the statistician (investigator). A code will be allocated to each patient and not revealed before the analysis has been completed. Once the analysis has been completed, the group to which participants had been allocated will be revealed. Due to the nature of this study, where the participants will be aware whether they will have access to the educational video, it is not possible to achieve blinding for the participants.
InterventionParticipants in the intervention group will be given access to a 6-min educational video 5 days after discharge from the hospital. This will allow the participants to become settled at home or at a training centre to ensure their receptiveness to the intervention. The educational video will present dietary recommendations from the Kostholdshåndboken (English: The Diet Handbook) published by Helsedirektoratet [58]. The dietary guidelines recommend that malnourished older adults increase their energy and protein intake and provide examples of how older adults can adapt and integrate the recommendations into their preferences and daily mealtime routines [58]. As the participants in the intervention group will be required to access the educational video via their email or smartphone, the video will be made accessible individually through a link sent out by the first author. Through the video, the participants will be encouraged to watch the video several times, with, for example, their relatives or other health personnel. Since it will not be possible to personalise the intervention to each participant, the video information on food and nutrition is based on the following general principles:
1)The information should be simple and correct, according to Contento and Koch [59]; this means keeping the words simple, since the field of nutrition can be full of technical jargon.
2)The information is presented using the active voice rather than the passive voice, since this makes the text more personal [59].
3)The length of sentences varies but is generally kept short [59]. The text of the education manual has undergone several ‘translations’ and revisions to make it easier for people with different assumptions and needs for information to understand. To prevent confusion among patients and strengthen the study’s criterion validity, we collaborated with a nutritionist who provided daily information to the user group to ensure we used precise words and expressions. Furthermore, the face validity of the educational video was confirmed by two bed-ridden patients in the user group who read the script and gave oral and written feedback on its contents. The intervention patients in the feasibility study did not have problems understanding the content, and significant changes in nutritional intake were observed.
In making the video, we used visual effects and pictures of different products so that participants would be able to imagine other food options more easily. Product images were obtained following current legislation, and there are no conflicts of interest or collaboration between the producers of the product images and the study’s authors.
The control group will have no access to the video and will not receive other intervention dietary advice in relation to this study. The selected hospital performs nutritional screening of patients in connection with admission as a part of the standard procedure at the hospital but does not provide nutritional information brochures or similar information upon discharge. However, as a standard procedure at the selected hospital, malnourished patients can receive advice or follow-up from healthcare professionals such as nutritionists on request, regardless of the intervention. No concurrent treatment or intervention will take place during the trial.
To strengthen retention, the participants in the intervention group will get a text message each week for the first 3 weeks after discharge and be contacted by phone to reduce technical problems opening the link, etc.
At follow-up after 3 months, the participants in the intervention group will be asked if they have seen the video and if they can remember how many times they have seen it. At the end of the intervention period, the authors will meet to review that both participants in the intervention group have been given access to the intervention as planned and that the intervention has not been given to the control group. The education video will not be modified during the course of the study.
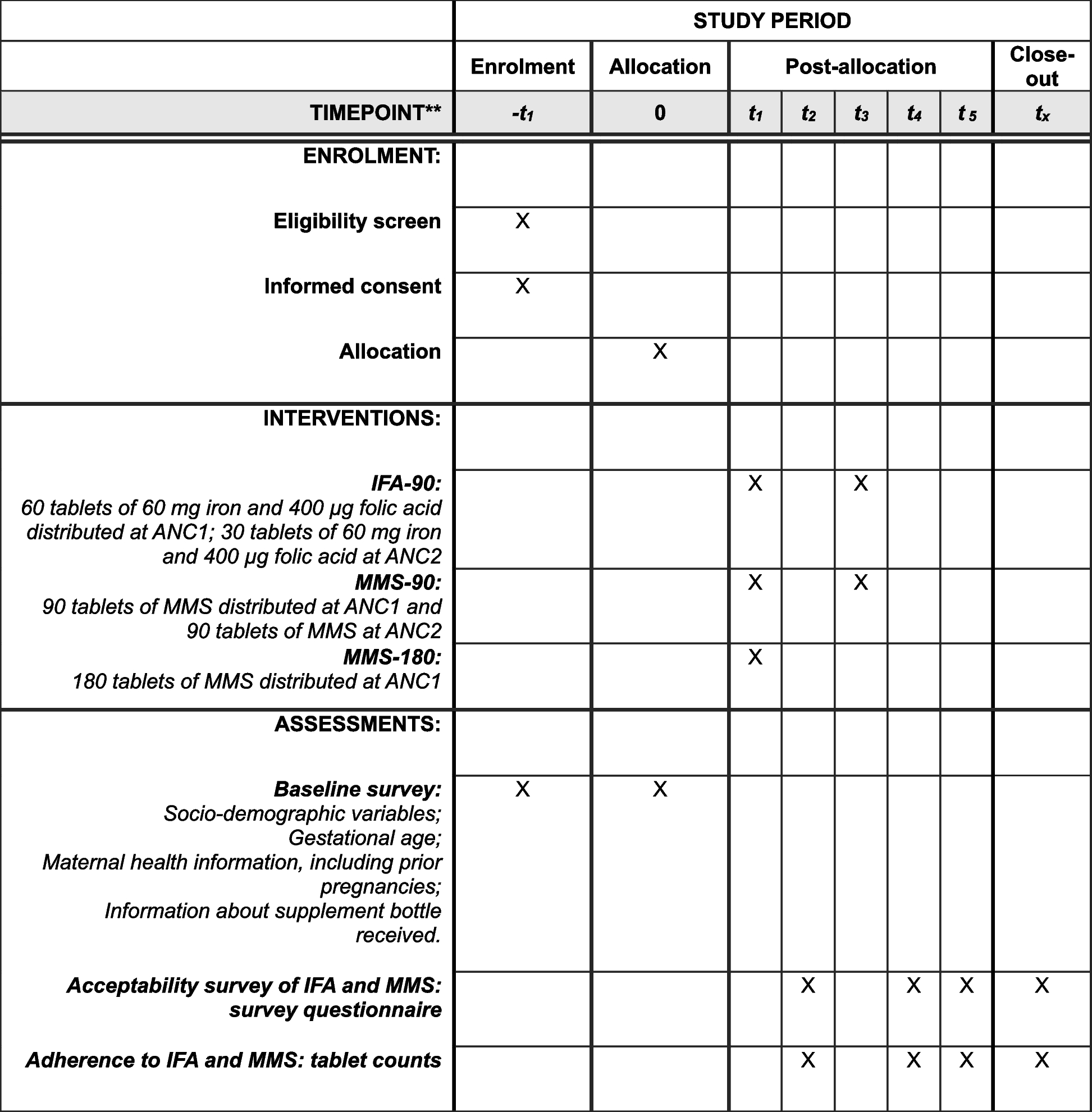
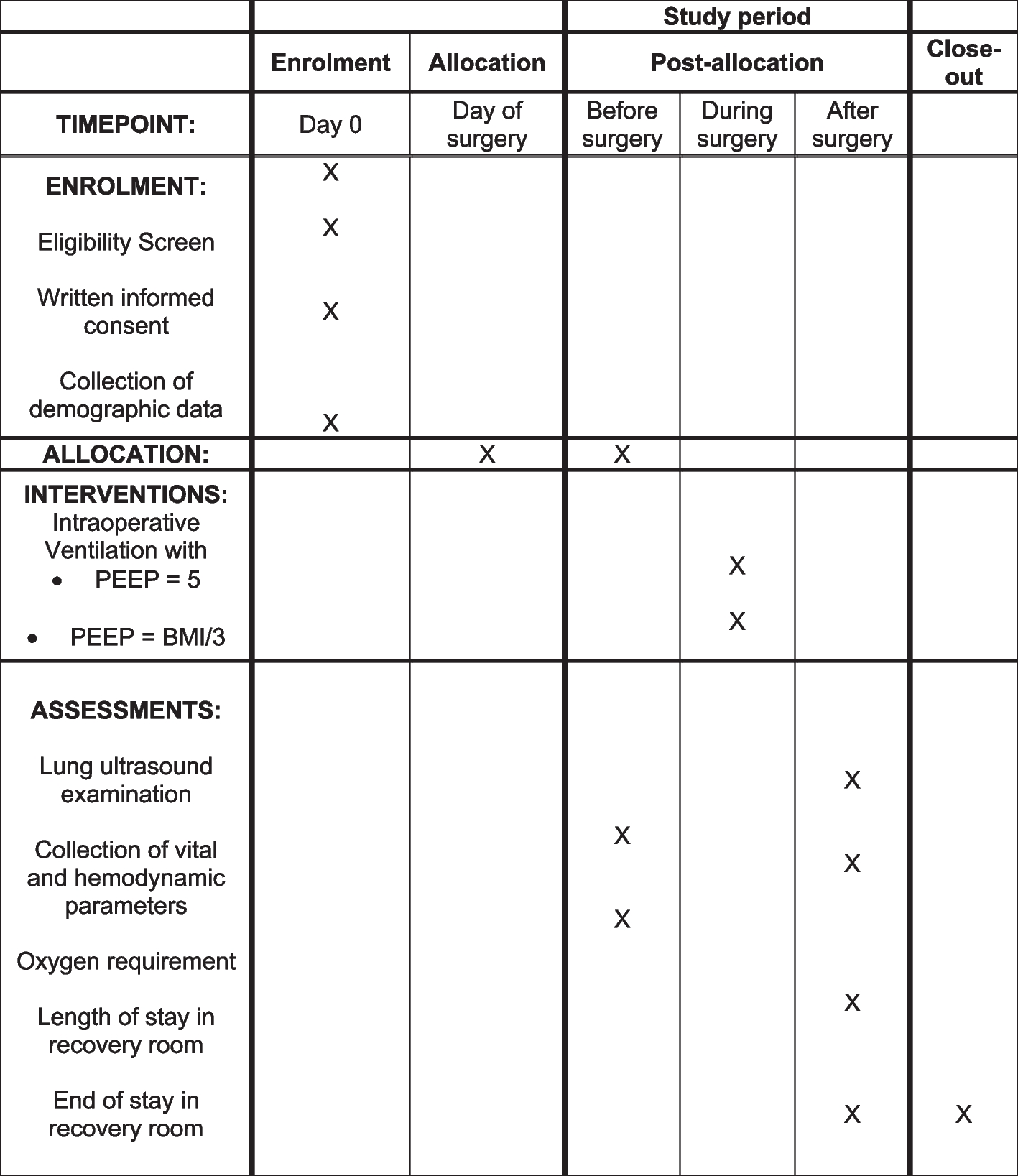
MeasurementsThe measurements will mainly focus on basic information, health-related quality of life, body composition measurements and readmission to hospital. The same externally trained data collector will carry out all measurements during the patients’ hospital stays and 3 months after discharge. We will register the number of readmissions within 3 months after discharge from the hospital. Figure 1 shows the schedule for enrolment, the intervention and the assessment of the trial according to the SPIRIT template.
Fig. 1 Demographic and clinical information collection
Demographic and clinical information collectionThe patients’ demographic information, including name, age, email address, phone number, level of education and need for home care, will be collected at the baseline in the hospital. Information on LOS and the number of readmissions will be collected from hospital records 3 months after discharge. This trial will not involve the collection of biological specimens for storage.
Primary outcomeHealth-related quality of lifeHealth-related quality of life will be assessed using the 36-item Medical Outcomes Study Short Form Questionnaire (RAND 36). The RAND 36 is grouped into eight health concepts using multi-item subscales that are combined into two main scales summarising physical and mental health. The physical health component correlates mainly with the subscales of bodily pain, physical functioning and role limitations on physical scales [60]. The mental health component correlates mainly with the three subscales of social functioning, mental health and role limitations on emotional scales [60]. The two subscales of general health and vitality have noteworthy correlations with both the mental and physical health components [60]. The participants’ answers will then be analysed and given a score following the recommendations of the RAND Corporation [61].
Secondary outcomeBody mass indexThe participants will be weighed upon admission to the hospital. Their weight will be measured in the morning, when they are wearing light clothes without shoes and using the electronic Withings Body Cardio WBS04; their weight will be recorded to the nearest 0.1 kg. To be sure that only the Withings Body Cardio weight will be used in all weight measurements, the use of bed weights will be prohibited during this project. If participants cannot stand on the scale, weight data will not be collected. Height will be estimated based on forearm length, counting the ulna length between the point of the elbow and the midpoint of the prominent bone on the wrist. This value will be compared to the values in a standardised high conversion chart from RxKinetics [62] and recorded to the nearest 0.5 cm. BMI will then be calculated based on participants’ weight (kg) divided by the square of their height (m) [63]. Values < 24 kg/m2 will indicate malnutrition.
The mid-arm circumferenceThe MAC will be measured using the midpoint between the olecranon and the acromion, with the arm extended, the muscles relaxed and the palm facing the thigh. The measurement will be recorded in centimetres, using the left hand. The participants will be classified as malnourished if the MAC is less than 24 cm and well-nourished if it is more than 24 cm [64].
Triceps skinfold thicknessThe TSF will be measured to estimate the fat mass in millimetres. A VirtuFits digital fat calliper will be used to measure the skinfold of the triceps of the left arm. The test will be repeated three times, and the average will be recorded.
The mid-arm muscle circumferenceThe MAMC will be calculated according to the following formula: MAMC (cm) = MAC (cm) − (3.14 × TFS × 0.1). Guided by Symreng [65], malnutrition will be indicated for women up to 79 years of age if the arm muscle circumference is less than 19 cm and for women over 79 years if the arm muscle circumference is less than 18 cm. Malnutrition will be indicated for men up to 79 years if the arm muscle circumference is less than 23 cm and for men over 79 years if the arm muscle circumference is less than 21 cm.
Handgrip strengthHandgrip strength will be measured using the Saehan Hydraulic Hand Dynometer and recorded to the nearest 0.5 kg for both the dominant and non-dominant hands. Following the manufacturer’s recommendations, the participant will sit with the shoulder adducted and naturally rotated, the elbow flexed at 90° and the forearm and wrist in a neutral position. The test will be repeated three times for each arm, and the average value for each arm will be recorded.
Knowledge of nutritionParticipants’ nutritional knowledge will be measured by questions asking them what they think would provide them with the most protein and energy among different kinds of food, such as apples, vegetables, meat, oat crackers and dairy products. For example, one of the questions is as follows: ‘Which of the following food categories do you believe is best for you to consume to ensure a good start to your rehabilitation: dairy products with little or a lot of fat?’ The inclusion group will be able to find answers to the questions through the educational video.
Readmissions after discharge from the hospitalReadmissions to the hospital will be measured for up to 3 months after discharge by each department’s internal data collector, who will have access to the department’s record system.
Data management and monitoringData analysisFor statistical analysis, SPSS version 28.01 (IBM Corp., Armonk, NY, USA) will be used. The statistical analysis plan will be based on the principles of the intention-to-treat approach. All ratio–interval scaled data will be tested for distribution using the F-test. When normally distributed, parametric statistical methods will be applied, such as Student’s t-test and the one-way analysis of variance (ANOVA). The data will be presented through means, standard deviation (SD) and 95% confidence limits. Normal or ordinal data will be presented as absolute numbers and frequencies. Differences will be determined using non-parametric tests (e.g. the X2-test, Mann–Whitney test and rank-sum test). The data from the RAND 36 questionnaire will be decoded using the SF-36 ordinal subscales and will be transformed into a ratio scale using the syntax SF-36 manual. The results will be presented from each of the eight subscales and the two main scales. The reliability of the SF-36 scales will be calculated using Cronbach’s alpha. When the data analysis is complete, the code regarding participants’ allocation to either the intervention or control groups will be revealed. Data will be collected as long as patients are included in the study. If patients withdraw their consent, data collection will stop. Deviation from the protocol will be registered and reported accordingly. Concerning post-assignment attrition, we will conduct an attrition analysis based on variables such as the participants’ gender, age, education level, marital status and help from home care. The attrition analysis will thus reveal any systematic loss at follow-up and identify whether there are participants with a higher likelihood of attrition.
MonitoringThe first author will continuously monitor the study in relation to registration. Specifically, (1) data will be exhaustively registered, (2) the number of potential participants who refuse to participate will be tracked, (3) participants who have viewed the education video will be contacted 1 week after they have received the video to check that they have been given access to it and (4) participants and their relatives will be contacted to ensure they have received information on how to contact the project management group in the event that they have questions or unwanted incidents occur.
Furthermore, a steering committee will be responsible for providing support for the study. Members of the steering committee will be from Nord University, leaders from the involved wards at the hospital and a user representative. The primary author will be the secretary for the steering group. The steering committee will meet at least monthly and receive reports of the study’s progress each week.
The first author will inform the steering committee if changes to the protocol occur, and any deviations from the protocol will be fully documented. Changes in the protocol will also be updated in the clinical trial registry. Since the research will be done at only one hospital and this is a low-risk intervention, a coordinating centre, project management group or data monitoring committee will not be needed.
Adverse event reporting and potential harmTo our knowledge, there is no evidence of adverse or serious events resulting from the intervention. There is no anticipated harm and compensation for trial participation. Still, we are aware that the participants belong to a vulnerable elderly population, and they will receive contact information from the first author and be encouraged to make contact if unwanted incidents occur. Supposing an unexpected serious event should nevertheless occur, this will be reported to the relevant regulatory bodies (sponsor and hospital) as required, and such reports will indicate expectedness, seriousness, severity and causality within one working day following the project management group’s awareness. Interim analysis and stopping rules are not needed because this is a low-risk intervention.
留言 (0)