Study setting
This single-center trial is recruiting participants at the inpatient and outpatient unit of the Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine Charité – Universitätsmedizin Berlin. All visits throughout the study will take place either in the study center (visit 1) or on the ward (visit 2: oral food challenge).
Eligibility criteria
Infants and children aged 8 months to 4 years with a sensitization (specific IgE (sIgE) ≥ 0.1 kU/l and/or skin prick test (SPT) wheal size ≥ 3 mm) against cow’s milk, hen’s egg, peanuts, and/or hazelnuts (target allergenic foods) and an already scheduled oral food challenge at the Charité – Universitätsmedizin Berlin (3–6 months after enrollment) for routine diagnostics are eligible for the trial. Infants are excluded from the study if they have previously consumed the target allergenic food they are sensitized to in relevant amounts. Further exclusion criteria are wheat allergy (study product contains wheat), severe other health issues (e.g., heart disease), a twin sibling already taking part in the study, and participation in another interventional trial.
Who will take informed consent?
The legal representative(s) of all participants must read, sign, and date the informed consent form before entering the study or undergoing any study-specific procedures. Before consent is given, the investigator or his/her representative will explain verbally the aim, method, source of funding, and the anticipated benefits and potential risks of the study to the parents; answer all questions regarding the study; and document the informed consent process.
Additional consent provisions for collection and use of participant data and biological specimens
As opt-it consent, included in the main study consent form, biological specimens are collected and partially stored for future analysis.
InterventionsExplanation for the choice of comparators
For eligible children (with sensitization against food allergens with unknown clinical relevance), an OFC is recommended in routine care. Until the OFC is conducted, the corresponding foods have to be avoided. This procedure is the current standard of care and corresponds to the recommendations in the placebo group of this trial.
Intervention description
In the active group, subjects consume daily 3 g of a sugar-free rusk-like biscuit powder, which contains small amounts of cow’s milk, hen’s egg, peanuts, and/or hazelnuts separately, corresponding to their sensitization(s). Therefore, a child may receive only one or up to four foods, each offered in a separate study powder. The allergen dose in each study product will be around 2 mg of food protein (Table 1).
Table 1 Allergen amount in study productThe sugar-free, rusk-like biscuit powder (3 g of powder/day using a measuring spoon) will be mixed with water, and optionally fruits, to a puree. The study product should be eaten daily throughout the study duration. The first feeding of the study product at visit 1 (V1) will be performed under medical supervision at the study center. The duration of the intervention is determined by the date of the routinely scheduled OFC at visit 2 (V2) and should be at least 3 months and up to 6 months after inclusion to the trial. In case of a postponed OFC (e.g., due to illness of the child), the duration of the intervention can be extended up to a maximum of 9 months.
In the placebo group, subjects consume daily the same sugar-free rusk-like biscuit powder, however, without cow’s milk, hen’s egg, peanuts, or hazelnuts following the same instructions as the active group. To ensure blinding, the first feeding of the placebo study product will also be performed under medical supervision at the study site.
In infants, first, weaning foods (e.g., vegetables) should have been successfully introduced for at least 1 week before feeding the study product. Mothers will be encouraged to continue breastfeeding while introducing weaning foods. Parents of both groups will be advised to avoid cow’s milk, hen’s egg, peanuts, hazelnuts, or their products in their child’s diet according to the individual sensitization profile.
Criteria for discontinuing or modifying allocated interventions
If the subject is unable to consume the recommended amount of rusk-like biscuit powder, e.g., due to feeding difficulties when weaning foods are introduced, the investigator and study physician may adjust the amount and/or dosing regimen. The investigators will prematurely discontinue the study intervention for a subject in case of significant health risk, i.e., after severe adverse events related to the study intervention requiring intensive care treatment, or non-compliance. In case of an intervention with more than one allergen, the intervention will be performed with separate study products. Therefore, in case of study product-related adverse events, intervention may only be discontinued of the corresponding product, whereas the other product(s) could still be offered.
Strategies to improve adherence to interventions
The parents of the study participants will be asked to maintain a weekly (e-)diary from V1 onwards to document consumption and any adverse events. In case of e-diaries, data are stored directly in a central database REDCap. Furthermore, patients will be called once during the study (phone call (PC)) to investigate compliance and tolerability of the regimen. They will be instructed to contact the investigation site by phone in the case of any objective immediate-type allergic reaction occurring within 2 h after food consumption including accidental allergic reactions to other food allergens. Also at V2, compliance, tolerability, and the entries of the (e-)diary will be reviewed and discussed with the investigator or delegated study staff.
Relevant concomitant care permitted or prohibited during the trial
All participants may continue their usual medications, as well as those taken for any concomitant diseases including wheezing and eczema throughout the study. All subjects who will undergo an oral food challenge at V2 will be advised to discontinue oral antihistamines 3–5 days before this procedure.
Provisions for post-trial care
Participants who developed a clinically relevant food allergy after completion of the trial will receive individualized dietary counseling. Moreover, parents are offered to contact the outpatient clinic of the Department of Pediatric Pulmonology, Immunology, and Critical Care Medicine, Charité – Universitätsmedizin Berlin for further routine consultation.
Outcomes Primary endpoint
The primary endpoint is IgE-mediated food allergy to the target food (hen’s egg, cow’s milk, peanuts, or hazelnuts), after 3–6 months of intervention (plus max. 3 months in case of postponed oral food challenge). For polyvalent-sensitized children, a “primary target food allergen” will be determined randomly at enrollment which will be used to define the primary endpoint.
The presence of IgE-mediated food allergy is defined by either of the following (Table 2):
Positive oral food challenge (plus sIgE ≥ 0.1 or positive skin prick test)
An immediate-type allergic reaction due to an accidental exposure (plus sIgE ≥ 0.1 or positive skin prick test)
Table 2 Diagnosis of food allergy for the determination of endpointsOFC, sIgE determination (blood sampling), and skin prick test (SPT) are performed onsite on the day of the scheduled visit (V2), which should be 3–6 months (plus max. 3 months in case of postponed oral food challenge) after the start of the intervention (V1). Specific IgE and/or SPT at baseline must be assessed no longer than 3 months prior to enrollment.
Secondary endpoints
The following secondary endpoints will be assessed after 3–6 months (plus max. 3 months) of intervention (V2):
1.
IgE-mediated food allergy determined by OFC (plus sIgE ≥ 0.1 or positive SPT) to the “primary target food allergen”
2.
IgE-mediated food allergy determined by an immediate-type allergic reaction due to accidental exposure (plus sIgE ≥ 0.1 or positive SPT) to the “primary target food allergen”
3.
IgE-mediated food allergy determined by OFC (plus sIgE ≥ 0.1 or positive SPT) or an immediate-type allergic reaction due to an accidental exposure (plus sIgE ≥ 0.1 or positive SPT) to either of the following (for children sensitized to the respective allergen at baseline):
(a)
Hen’s egg
(b)
Cow’s milk
(c)
Peanuts
(d)
Hazelnuts
4.
Occurrence (frequency and severity) of immediate type allergic symptoms
5.
Occurrence of gastrointestinal symptoms
6.
Multiple food allergies
7.
Number of food allergies
8.
Change to baseline of SCORAD (in subjects with eczema)
9.
Change to baseline of EASIscore (in subjects with eczema)
10.
Wheezing
11.
Allergen-specific IgE to hen’s egg, cow’s milk, peanuts, and/or hazelnuts
12.
Wheal size measured by skin prick testing to hen’s egg, cow’s milk, peanuts, and/or hazelnuts
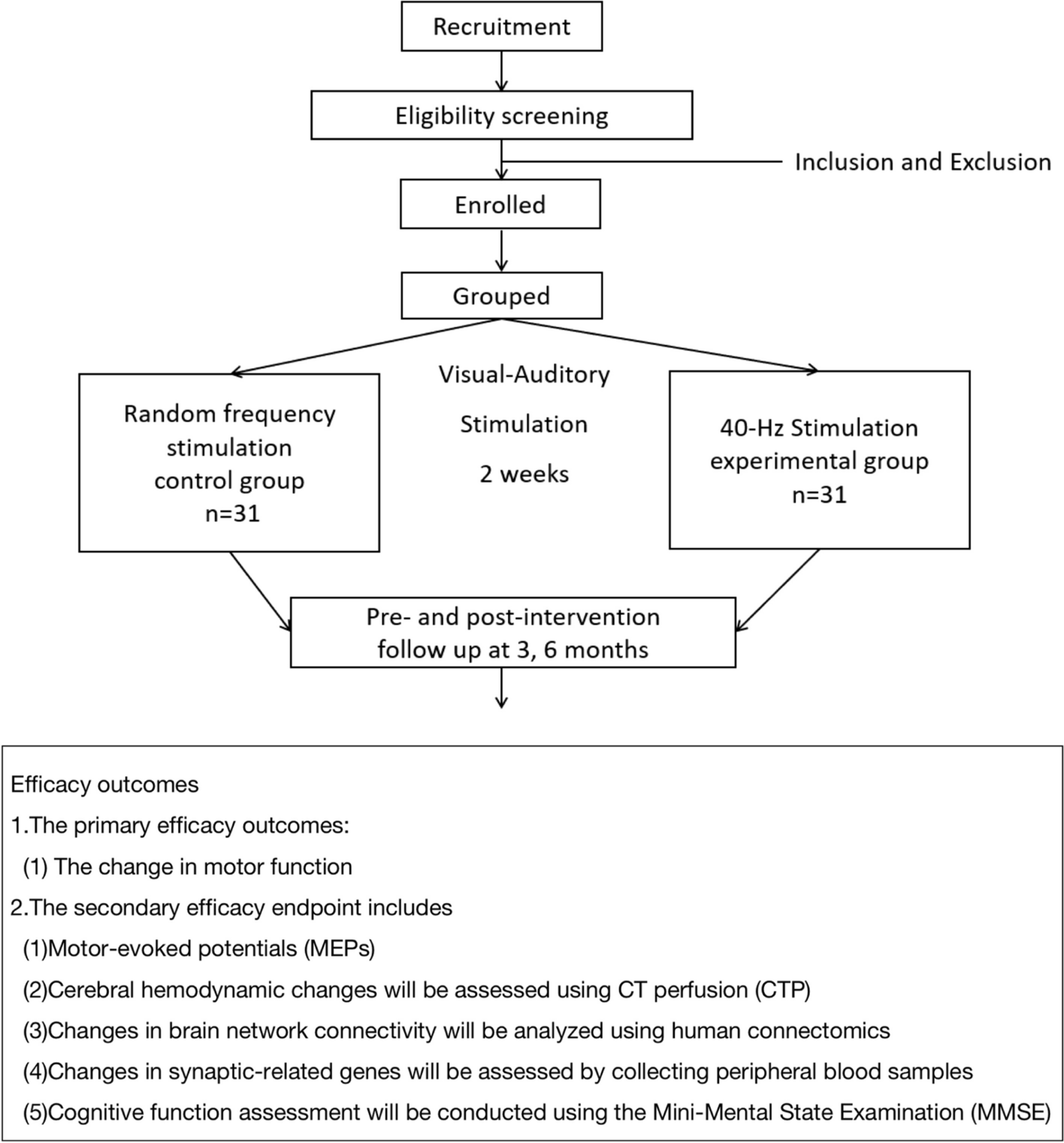
Participant timeline
During the screening visit (V1), the participation criteria will be checked, and information on demographics, subject/family characteristics, relevant medical history, medication, and nutrition will be recorded. In addition, anthropometric measurements and a physical examination will be performed including the assessment of the severity of eczema (in case of eczema). Skin swabs and stool and saliva samples will be collected. Blood will be collected, or a skin prick test (SPT) will be performed, if the child’s sensitization status was assessed ≥ 3 months prior to V1. Transepidermal water loss (TEWL) will be measured, and palmar hyperlinearity will be determined. Parents are asked to collect a dust sample from the bed and living room within 24 h prior to both study visits. Eligible children will be randomized to one of the two study groups. The first feeding of the study product will be performed at the study site. The study product will be dispensed. Parents will be asked to fill out a (e-)diary for the entire study period (from V1 to V2). Two weeks after V1, parents will be called (PC1) to review compliance, tolerance of the study product, and adverse events. The study product will be consumed daily until an oral food challenge in routine diagnostics at V2 will be performed after their individual waiting time of 3–6 months. At V2, information on medical history, medication, and nutrition will be recorded. In addition, anthropometric measurements and a physical examination will be performed. Skin swabs and stool and saliva samples will be collected. TEWL will be measured. A blood sample will be collected, and a SPT with cow’s milk, hen’s egg, peanuts, and hazelnuts will be performed. Oral food challenges will be performed according to standardized procedures in a double-blinded placebo-controlled manner (Table 3).
Table 3 Study visits and proceduresSample size
A total of 138 children are planned to be included in the trial. Analyzing 124 children (62 in each group) will result in at least 80% power assuming a difference in the primary endpoint (food allergy after 3–6 months (plus max. 3 months) of 35% in the active group vs. 60% in the control group, based on a two-sided chi-squared test with significance level 5% (two-sided). To compensate for a potential dropout rate of about 10%, 138 infants will be randomized (69 children in each group). Sample size calculation was performed in SAS for Windows version 9.4 (SAS Institute, Cary, NC, USA).
Recruitment
Recruitment of participants will take place at the inpatient and outpatient unit of the Department of Pediatric Respiratory Medicine, Immunology, and Critical Care Medicine of the Charité – Universitätsmedizin Berlin. All parents of children with a scheduled OFC with cow’s milk, hen’s egg, peanuts, and/or hazelnuts who have not previously consumed the food they are sensitized to in relevant amounts will be considered for participation. Additionally, we aim to approach participants via flyer advertisement, advertisement in journals, magazines, and social media as well as at information events for (prospective) parents.
Assignment of interventions: allocationSequence generation
After enrollment performed by the study investigators, participants will be randomly assigned to receive verum or placebo. The randomization will be prepared by the trial statistician and will be implemented within the REDCap database system by the data manager. Children will be randomized to receive verum or placebo (1:1 allocation ratio) for each food allergen against which the child is sensitized.
Randomization will be performed in blocks (with varying block lengths) and will be stratified by the following:
Primary allergen (cow’s milk, hen’s egg, peanuts, or hazelnuts)
Age (≤ 2 vs. > 2 years of age)
Grade of sensitization to the primary food allergen (low vs. medium vs. high) defined by either of the following:
◦ Specific IgE levels of the primary food allergen, according to the following CAP classes:
Low: IgE ≥ 0.10 ≤ 3.50 (CAP 0–2)
Medium: IgE > 3.50 ≤ 50.0 (CAP 3 and 4)
High: IgE > 50.0 (CAP 5 and 6)
◦ According to skin prick test results of the primary food allergen (in case of missing specific IgE values)
Low: wheal size ≥ 3 mm ≤ 5 mm
Medium: wheal size > 5 mm ≤ 9 mm
High: wheal size > 9 mm
Planned duration of intervention, determined by the date of the routinely scheduled OFC (< 5 vs. ≥ 5 months)
Concealment mechanism
Allocation of subjects will be performed concealed (i.e., without knowledge of group allocation) using REDCap where each participant will be allocated to 1 out of 20 kit codes for the corresponding allergen(s) for the interventional product (10 codes active, 10 codes placebo).
Implementation
After enrollment performed by the study investigators, participants will be randomly assigned to receive verum or placebo. The study product will be labeled with the kit codes by staff, who is unblinded, but not otherwise involved in the study.
Assignment of interventions: blindingWho will be blinded
Study investigators, all involved study staff, participants, and their parents are blinded to whether the participants will receive allergen-containing rusk-like biscuit powder or allergen-free rusk-like biscuit powder (placebo powder). The main (confirmatory) statistical analysis will be performed blinded to treatment allocation. Unblinding per subject will be performed at the end of the study.
Procedure for unblinding if needed
In case of serious allergic reactions suspected to be related to the study intervention, unblinding will be performed at the discretion of the investigator. To ensure the unblinding process, emergency envelopes, providing information on the individual interventional group the subject is assigned to, are stored at the study center. Once unblinding has been performed, it will be documented in the source data, reported to the PI, and will result in discontinuation of intervention.
Data collection and managementPlans for assessment and collection of outcomes
Assessment and collection of data and biosamples will be performed by the trained study staff according to standard operating procedures (SOPs). During V1, the participation criteria will be checked and information on demographics, subject/family characteristics, relevant medical history, medication, and nutrition will be recorded. In addition, anthropometric measurements and a physical examination will be performed including the assessment of the severity of eczema by SCORAD [12] and EASIscore [13] in case of eczema. Skin swabs and saliva samples will be collected. Skin swabs are collected and preserved in DNA/RNA shield collection tubes containing medium (DNA/RNA Shield; Zymo Research, Irvine, CA, USA). Saliva samples are collected both with Zymo Swabs as well as with Salimetric Swabs (SalivaBio Swab; Suffolk, Great Britain). The investigator will instruct the parents to collect stool samples (using OMNIgene-GUT tubes, OMR-200; DNA Genotek, Ontario, Canada) from the infants at home. Blood will be collected, or a SPT (all extracts including positive and negative controls: ALK Abelló, Germany) will be performed, if the child’s sensitization status was assessed ≥ 3 months prior to V1. Transepidermal water loss (TEWL) measurement will be performed with the Tewameter TM Hex (Courage + Khazaka Electronic GmbH, Germany). Palmar linearity pattern will be analyzed and documented by palm photographs. The parents should further collect a dust sample using dustream® collector DU-ST-1 (Indoor Biotechnologies LTD, Cardiff, UK) from the child’s bed (or the parent’s bed, if the child sleeps in this bed four nights or more) and the living room. At V2, information on medical history, medication, and nutrition will be recorded. In addition, anthropometric measurements and a physical examination including SCORAD [12] and EASIscore [13] in case of eczema will be performed. Skin swabs and stool and saliva samples will be collected. TEWL will be measured. Moreover, a blood sample will be collected, and a SPT with hen’s egg, cow’s milk, peanuts, and hazelnuts will be performed. OFC will be performed in a double-blinded, placebo-controlled manner in accordance with the clinical routine practice of the Charité – Universitätsmedizin Berlin, based on the PRACTALL international guidelines for OFC and stopped using standardized stopping criteria based on PRACTALL guidelines [14]. Roasted, defatted peanut flour; defatted hazelnut flour; fresh cow’s milk; and pasteurized raw hen’s egg will be used for OFC. Up to seven increasing dose steps at 30-min intervals using a semi-log scale ranging from approximately 2 mg to 3 g food protein (depending on the individual allergen) will be administered. In case of absence of any objective, allergic symptoms, a cumulative dose of 4.4 g of food protein will be administered on another day. To patients with allergic reactions to raw hen’s egg, an additional food challenge with baked hen’s egg will be offered.
Plans to promote participant retention and complete follow-up
Two weeks after V1, parents will be called (PC1) to investigate compliance and tolerability of the study product. In case of discontinuation of the intervention or deviation from the study protocol, participants are encouraged to maintain the scheduled visits or phone calls, and collection of all possible data is planned.
Data management
Research Electronic Data Capture (REDCap) will be used as an electronic case report form (eCRF) to collect and manage the study data. REDCap is hosted at the Charité – Universitätsmedizin Berlin and provides an interface for data entry for clinicians. An audit trail will be integrated for tracking data entries and corrections. Data access and storage will follow the data security concept of the Charité including firewalls on the campus level, institutional level, and individual computer level and password-protected access to all computers and folders, which contain sensitive data.
Confidentiality
Participant’s privacy and confidentiality will be respected throughout the study. To ensure the protection of personal data, the national legal requirements including the EU General Data Protection Regulation (GDPR) regarding data confidentiality will be followed. Appropriate consent for collection, use, disclosure, and/or transfer (if applicable) of personal information must be obtained in accordance with local data protection laws. A unique participant identifier will be allocated to each participant and assigned chronologically prior to proceeding with study screening. These participant identifiers rather than names will be used to collect, store, and report participant information, including documentation in the eCRF. If the participant’s name appears on any other document (e.g., laboratory report), it must be obliterated on the copy of the document to be, e.g., uploaded to the eCRF. The investigator must retain records and documents, including signed informed consent forms, pertaining to the conduct of this study for 15 years after study completion and final publication. No records may be destroyed during the retention period without the written approval of the sponsor.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use
Biosamples obtained in this clinical trial will enable the mechanistic sub-projects of the CRU to investigate the mechanism of food allergy and tolerance development within the clinical research unit. In particular, the role of the gastrointestinal as well as skin microbiome, the IgEome, genetic and epigenetic mechanisms, antigen-specific immunologic reactivities as well as inflammatory circuits, and serological biomarkers will be investigated. The methods of these sub-projects will be described elsewhere. Only the participant identifier will be used to collect and store biosamples. Biological specimens like house dust samples or saliva may be partially stored at the Charité – Universitätsmedizin Berlin.



留言 (0)