
記住我
Successful implantation of an embryo into the endometrium requires the specification of extra-embryonic and embryonic lineages that will give rise to the placenta and the embryo itself. As a result of cleavage, compaction, radial polarization and asymmetric divisions of the newly formed zygote, two cell lineages of different developmental potential arise with an external outer cell layer, the trophectoderm, possessing epithelial characteristics enclosing a population of non-epithelial cells, the inner cell mass (ICM) (1). Epithelial cells are characterized by an apical membrane confronting the external environment, lateral membranes contacting neighboring cells and basal domains anchored to a basement membrane interacting with the extracellular matrix (1). This kind of organization subdivides cells, and thus tissues, morphologically and functionally into different compartments, enabling maintenance of an ion- and size-selective diffusion barrier, cell shape, cellular adhesion, communication, and cytoplasmic/surface polarity, which are all essential for intracellular machinery (2–4). Likewise, the outer trophectodermal cell layer, an epithelium analogue, serves as a chemical barrier that separates the embryo from the external uterine environment, maintaining blastocyst integrity, developmental potential and viability. This review focused on the epithelial characterization of the pre-implantation embryo and the structure, biogenesis and developmental functions of intercellular junctions that are essential for developing such phenotype. Functionally, intercellular junctions can be classified into three categories: 1-Anchoring junctions divided into adherens junctions (AJ), desmosomes, hemidesmosomes and focal adhesions; 2-Tight junctions (TJ), and 3-Gap junctions (GJ) (4) (Figure 1). Individual roles of all of these junction types concerning trophectoderm maturation and key events of embryo development will be emphasized separately. By assembling our current understanding of junction biogenesis during embryonic development, we aim to support studies on assisted reproductive technologies and embryo selection for IVF.
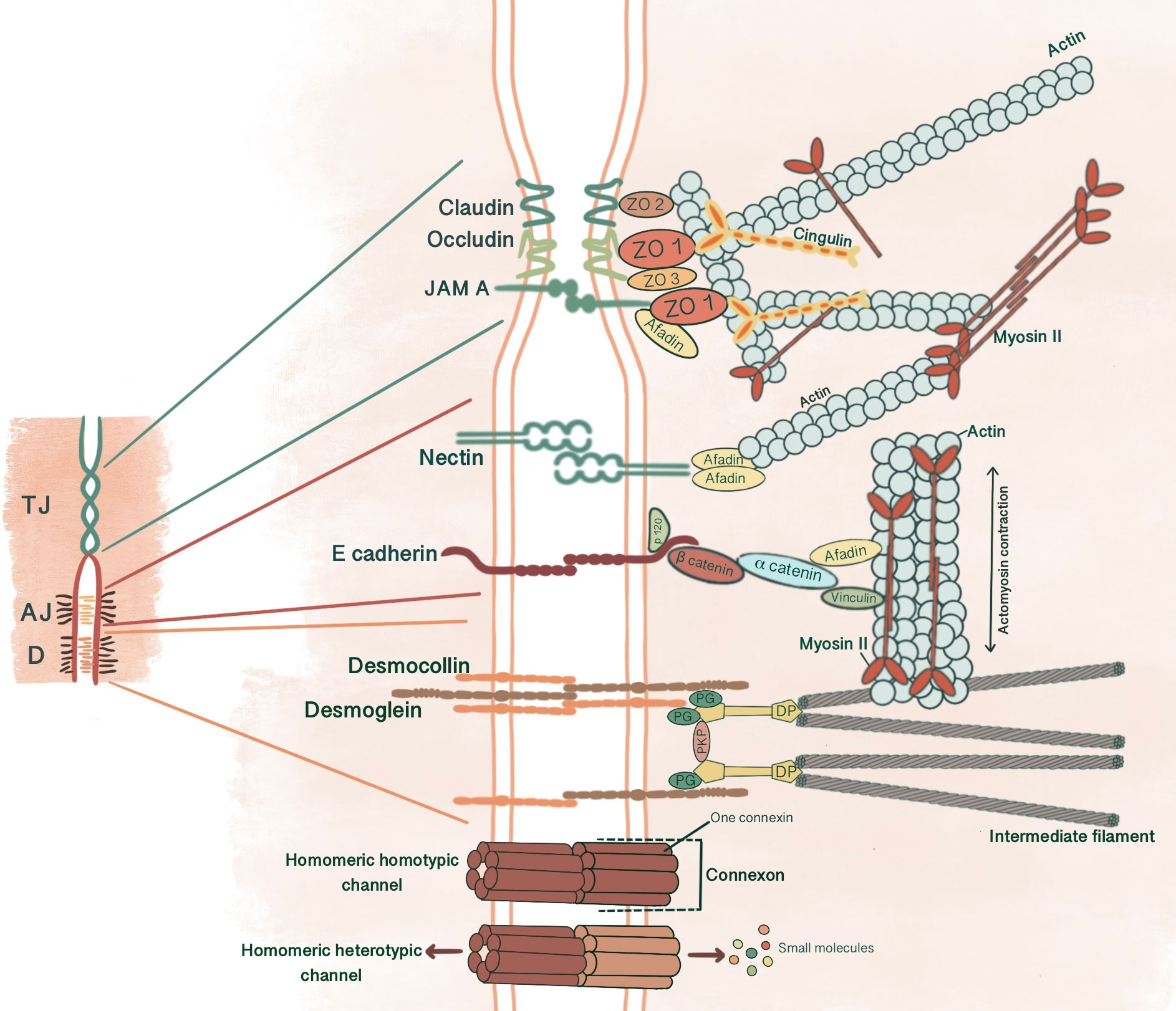
Figure 1 Simplified schematic presentation of basic structural components and molecular composition of intercellular junctions. (TJ, Tight junctions; AJ, Adherens junctions; D, Desmosomes; ZO, Zonula occludens; PG, Plakoglobin, PKP, Plakophilin; DP, Desmoplakin).
2 Adherens junctionsAJs are cell-cell adhesion complexes that form extracellular adhesive contacts between cells to maintain tissue cohesion, sense and respond to tensile forces at the contact interface, establish cell polarity and form intracellular links to cytoskeletal elements (5, 6). AJ’s ability to localize proteins to subcellular compartments allows modulation of signaling pathways (7). AJs comprise three main components: transmembrane cadherins, armadillo family members and cytoskeletal adaptor proteins. This core cadherin-catenin complex binds to actomyosin cytoskeleton and signaling proteins, influencing the overall mechanobiology of cells.
Depending on the tissue types and developmental stages, different AJ conformations might be present along the cell-cell interface. These variants of AJ such as linear AJ, focal AJ, zonula AJ (ZA), tricellular AJ and fascia AJ differ in their molecular composition, organization of the associated actomyosin skeleton and stability (6). Newly forming AJs are discontinuous cellular adhesions, characterized by a spot-like, punctate appearance. In maturing epithelial cells, the apical junctional complex contains ZA junctions underneath the apical TJ, forming a tight belt-like structure that links cells into continuous sheets, creating highly polarized cells with separate apical and basolateral membranes (6, 8).
Cadherins initiate cellular adhesion through forming trans-dimers with adjacent cadherins via their most distal extracellular domain in a calcium-dependent manner (6). The cadherin family consists of type 1 cadherins (e.g. E- cadherin expressed broadly in epithelia, P-, N-, M-, R-cadherin), type 2 cadherins (e.g. VE-cadherin restricted to vasculature), desmosomal cadherins (desmocollin and desmoglein) and subfamily of cadherin-like molecules (9). E-cadherin, a type of classical cadherin, is a single-pass transmembrane protein comprising five extracellular cadherin domains which are bound together by Ca+2 ions, a transmembrane domain and a C-terminal cytoplasmic domain (6, 10). The intracellular domain consists of a juxtamembrane domain (JDM) that binds p120-catenin and α-catenin binding domain (CBD) which binds β-catenin (5, 10). Catenins are cytoplasmic proteins that allow interaction of cadherin complex with the cytoskeletal elements. Catenins contribute to cadherin function in three ways: enabling direct physical link of cadherins to actin cytoskeleton, regulating signaling to cytoskeleton by tyrosine kinases and small GTPases, controlling the adhesive state of the cadherin extracellular binding domain (11). β-catenin consists of an amino-terminal region, a central domain of 12 armadillo arm repeats and a carboxy-terminal (10). p120-catenin contains 9 arm repeats preceded by an amino terminal sequence that varies in length, creating four splice varients (10). p120 catenin association with E-cadherin is essential in the formation of stable cell-cell adhesions. In cells expressing p120-uncoupled E-cadherin, there is a failure in the formation of continuous circumferential actin ring and insertion of these actin cables into peripheral concentrations of E-cadherin. This causes a failure in transit from loose to tighter cell-cell adhesions (12). α-catenin, the link between the cadherin-catenin complex and the cytoskeleton, contains 1) an N-terminal domain that binds β-catenin and plakoglobin 2) a central modulatory domain that binds vinculin, α -actinin; 3) a C-terminal domain that binds ZO-1, F-actin and α1-helix. Thus, α-catenin acts as the mechanosensor and mechanotransducer that goes under conformational changes in response to force and regulates actin cytoskeleton in a tension-dependent manner (6, 11). In addition, α-catenin is responsible in maintenance of junction stability. α-catenin suppressed cells lose their cell-cell contacts and demonstrate disrupted localization of β-catenin, E-cadherin and TJ protein ZO-1 (13) (Figure 1).
E-cadherin, α-catenin and β-catenin mRNA are all present in unfertilized eggs, 2-cell, 8-cell and blastocyst stages, with a decrease at the 2-cell stage. Maternally derived E-cadherin mRNA and protein is present in unfertilized egg and 1 cell-stage embryo, assembled into a protein complex with catenins to enable adhesive interactions between oocyte and cumulus cells (14). However, de novo E-cadherin transcription from the embryonic genome starts at the late 2-cell stage. With immunoelectron microscopy, 2- and 4-cell stage embryos demonstrate an even distribution of E-cadherin on cell surface of blastomeres. At the 4-cell stage, E-cadherin concentration increases in close membrane appositions. Following the 8-cell stage and compaction, E-cadherin starts accumulating at cell contact sites, removed from the apical membrane domain. In 16- and 32-cell stages, E-cadherin is redistributed in cells committed for epithelial cell differentiation and becomes basolaterally localized between adjacent cells in the outer cells of the morula and TE. Whereas, inner cells and ICM demonstrate an even distribution along the cell membrane (15). LIMK is essential for early cleavage compaction through AJ assembly and actin filament organization. LIMK1/2 knockdown in porcine embryos causes abnormal cleavage, reduced blastocyst formation, disrupted localization of β-catenin, E-cadherin, ZO-1, CXADR and decreased cortical actin levels (16). The basolaterally distributed E-cadherin associates with catenins that in turn connect with cytoskeletal structures. This cytoskeletal anchorage of the cadherin-catenin complex to actin influences the strength of adhesiveness of E-cadherin (17). With immunofluorescence microscopy, E-cadherin-catenin complex demonstrates a strong membranous localization at cell-cell contact sites in all development stages of mouse embryo, including 2-cell stage. (18). Rac 1, a small GTPase, is localized adjacent to cell membranes of 2- and 4- cell stage blastomeres and shifts to the cytoplasm with compaction. Additionally, Natale et al. Suggested that Rac-1 is an important regulators of E-cadherin-catenin complex during murine preimplantation embryonal development (19). β-catenin is detected in the surface and the pronuclei of zygotes, whereas at the surface of blastomeres of 2- and 8-cell embryos. (20). So, at the 2-cell stage, these adhesion proteins already form a complex and are localized to cytoskeleton-bound membrane domains to enable cell-cell contact between blastomeres. In immunofluorescence evaluation of the mouse embryos, total and active (dephospho) β-catenin is expressed in all stages of preimplantation development, starting from the 1-cell stage to blastocyst. While before the morula stage, active β-catenin is mostly localized in nuclei of all embryonic cells, it is predominantly present in TE of blastocysts (21). After the hatching of fully expanded blastocyst from zona pellucida for implantation, the signal for active β-catenin disappears. On E6, β-catenin appears in invasive trophoblasts, followed by the embryo on day 7 (22). In human embryos, β-catenin protein is localized in the cortical region under the cell membrane at 6-cell, 8-cell stage embryos, and both TE and ICM of blastocysts (23).
E-cadherin (-/-) embryos show defects in molecular architecture involving ZA, TJ and cortical actin filament organization. In addition, α- and β-catenin expression levels are significantly reduced (24). Removal of the N-terminal part of β-catenin, that allows the binding of α-catenin and E-cadherin, in oocyte disrupts adhesion between individual blastomeres. In addition, E-cadherin mislocalizes to the cytoplasm in 2- and 4-cell stage embryos, which is reversed during 4- to 8- cell stage transition with paternal allele synthesis of β-catenin (20).
2.1 E-cadherinThere are four prominent roles of E-cadherin containing ZA in pre-implantation embryonic development: 1) Mediating compaction, 2) Triggering radial polarization by inducing PAR asymmetry via cell contact cues, 3) Cell fate determination by differential activation of the Hippo signaling pathway in inner and outer cells through E-cadherin & apical polarity complex association with AMOT, 4) Directing hydraulic fracturing during blastocoel formation by reorganization of E-cadherin at cell-cell contacts.
2.1.1 Mediating compactionEmbryo compaction, which is the first critical morphogenetic process that occurs during embryonic development is initiated at the 8-cell stage (25). Starting from cell contact points proceeding outwards, adjacent blastomeres flatten against each other, maximizing their contacts and generating a mass of cells with no distinguishable cell boundaries (26). Although the exact mechanism that drives compaction is still unknown, three primary processes were suggested to mediate this morphological change: increased cell-cell contact due to a change in cellular adhesive properties, E-cadherin dependent filopodia formation and actomyosin mediated increase in surface contractility (27). AJ links to the cell cytoskeleton elements like actin, myosin 1 and ezrin/radixin/moesin is essential in mediating cell shape changes during compaction by transmitting the main driving forces generated by the actin cytoskeleton to the cell surface (27). Ca+2 dependent cell-cell adhesion system generates the force necessary for early 8-cell compaction. E-cadherin redistributes contractility generated by actomyosin away from cell-cell contact points toward the cell surface, causing a twofold increase in tension at the cell-medium interface (28). When embryos are cultured in Ca+2 free medium, compaction is inhibited. In addition, when embryos that have already compacted in vivo are transferred to Ca+2-free medium, embryos uncompact within 30 minutes (26).
Embryos homozygously knockout for E-cadherin, although undergo compaction due to residual maternal E-cadherin, fail to form blastocyst cavity and show prenatal lethality (29). Embryos lacking maternal E-cadherin show a lack of adhesion in blastomeres until the late morula stage, when E-cadherin translated from paternal alleles reaches the blastomere surface. Compaction of embryos that lack maternal E-cadherin is enabled, although a cell division later than normal embryos, by adequate blastomere contact with confinement within the zona pellucida until paternal E-cadherin is synthesized (20). In the absence of both maternal and zygotic E-cadherin, embryos develop as loose aggregates of cells, never compact and do not show normal epithelial morphology in outer blastomeres (30).
Live mouse embryo imaging demonstrates filopodia containing E-cadherin, F-actin and Myo 1 extending onto neighboring cells, transmitting the cell-autonomously generated cortical tension. There is temporal coordination between filopodia extension-retraction and cell shape change. Cells that are relatively round before filopodia extension undergo elongation at their apical border and become rounded again before division by filopodia retraction. Knockdown of filopodia components such as E-cadherin, α- and β-catenin causes failure in compaction. Comparably, inducing filopodia formation by increased expression of Myo10 causes premature compaction of the embryo (31).
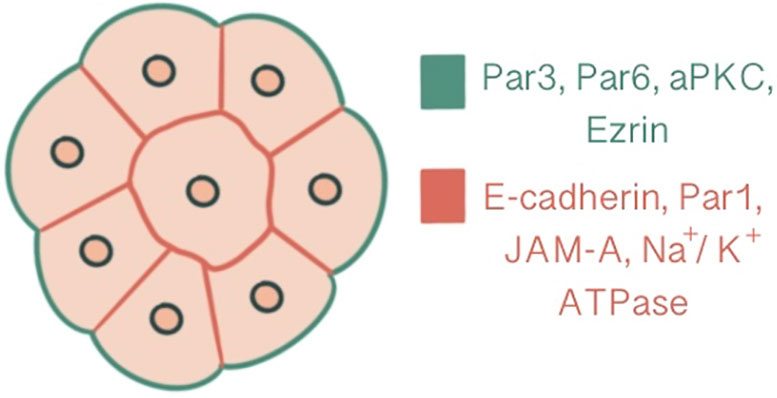
2.1.2 Triggering radial polarization by inducing PAR asymmetry via cell contact cuesIn parallel to compaction, radial polarity, the asymmetric localization of apical and basolateral polarity regulators, develops at the 8-cell stage as the second key morphogenetic event of the mouse embryonic development (1). The cell contact region where the junctional proteins will be recruited comprises the basolateral domain. In contrast, the contact-free surface enriched with microvilli, actin and actin-binding proteins marks the apical domain (32). The majority of cells become polarized in 6 hours within the generation of an 8-cell embryo, while some may start as early as 2 hours. The presence of cell contact is required to generate polarity, as the contact points guide the axis of polarization (33, 34). This polarity axis organizes cytoplasmic content and serves as a memory for generating two distinct cell types with differentially inherited polarized cellular domains (33). Subcellular machinery of specification of the polarized cell surfaces in embryos is regulated by spatial and temporal localization of apical and basolateral polarity proteins (35). Three main polarity regulator proteins ultimately scaffold Rho GTPase to specific membrane domains: the Par group at close localization to AJC and the primary cilium, the crumbs complex at the apical side of AJC and the scribble complex on lateral membranes below the AJC (1, 3, 36). Par3-Par6-aPKC is the core complex that regulates polarity in pre-implantation embryos. Par-6 binds to the amino terminus of Par-3 through its PDZ-1 domain. Par-6 associates with aPKC via direct head-to-head association and forms a complex that can associate with and phosphorylate Par-3 (37, 38). E cadherin is one of the first proteins to demonstrate a polarized distribution at cell contact sites with compaction, followed by Ezrin, Par6, Par3, aPKC at apical domain and Par-1, JAM-1, Na/K ATPase at basolateral domain (1, 39) (Figure 2). E-cadherin mutant embryos demonstrate an overlap between apical and basolateral proteins: PKCζ becomes localized throughout the majority of outer cell membrane, overlapping with Na/K ATPase β-1 subunit, Jam1 and Lgl1. In addition basolateral markers also accumulate as intracellular puncta and vacuole-like structures within the cell (30).
Figure 2 Polarization demonstrated at a 16-cell stage embryo, central slice. Ezrin, Par6, Par3, aPKC are present at the apical domain which is enriched with actin, whereas Par-1, JAM-A, Na/K ATPase and E-cadherin are localized at the basolateral domain.
In the mouse embryo, de novo polarization follows two steps: 1) Actomyosin localization to the cell-contact free surface during the early-to-mid 8-cell stage by PKC-RhoA activation through PLC-mediated PIP2 hydrolysis, 2) Apical enrichment of PAR proteins at mid-late 8-cell stage forming a mature apical cap, whereas F-actin redistribution to form a ring-like structure around the PAR enriched domain (40). Restriction of Par polarity proteins to contact free surface has two distinct effects: a developmental role in cell fate specification by modification of the Hippo signal pathway and an effect on tissue morphogenesis by directing cytoskeletal asymmetries (1, 37).
After polarization and compaction, the morula undergoes two sets of asymmetric divisions (from 8-to-16 cells and 16-to-32 cells), generating two distinct cell populations that occupy different positions within the morula (27, 41–;43). The inheritance of the apical domain, depending to the division angle of the embryo, determines the fate of daughter cells (43). Polar cells, which have an apical surface, become positioned peripherally while apolar cells are placed centrally within the embryo (43). This process of differential inheritance is the foundation of ICM and TE lineage differentiation (43). SPECC1 is detected at apical cell-cell contacts at the blastocyst stage, and knockdown of SPECC1 disrupts paracellular sealing, reducing the rate of blastocyst development (44). Recent studies showed division-independent routes, such as internalization, for inner cell allocation. Samarage et al. suggest that apical constriction of the cortical actomyosin network is the primary morphogenetic mechanism involved in the first spatial segregation of cells. In the 12-cell stage, constricting cuboidal shape daughter cells with high cortical tension decrease their apical surface and gradually position closer to the center of the embryo. Daughter cells demonstrating a lower tensile force remain outside and adopt a wedge shape. Although the junction length between constricting cells and their neighbors decreases, E-cadherin levels or mobility do not differ among cells during apical constriction. Myosin II distribution heterogeneity has a more specialized role in inner cell allocation rather than E-cadherin-mediated cell adhesions (45).
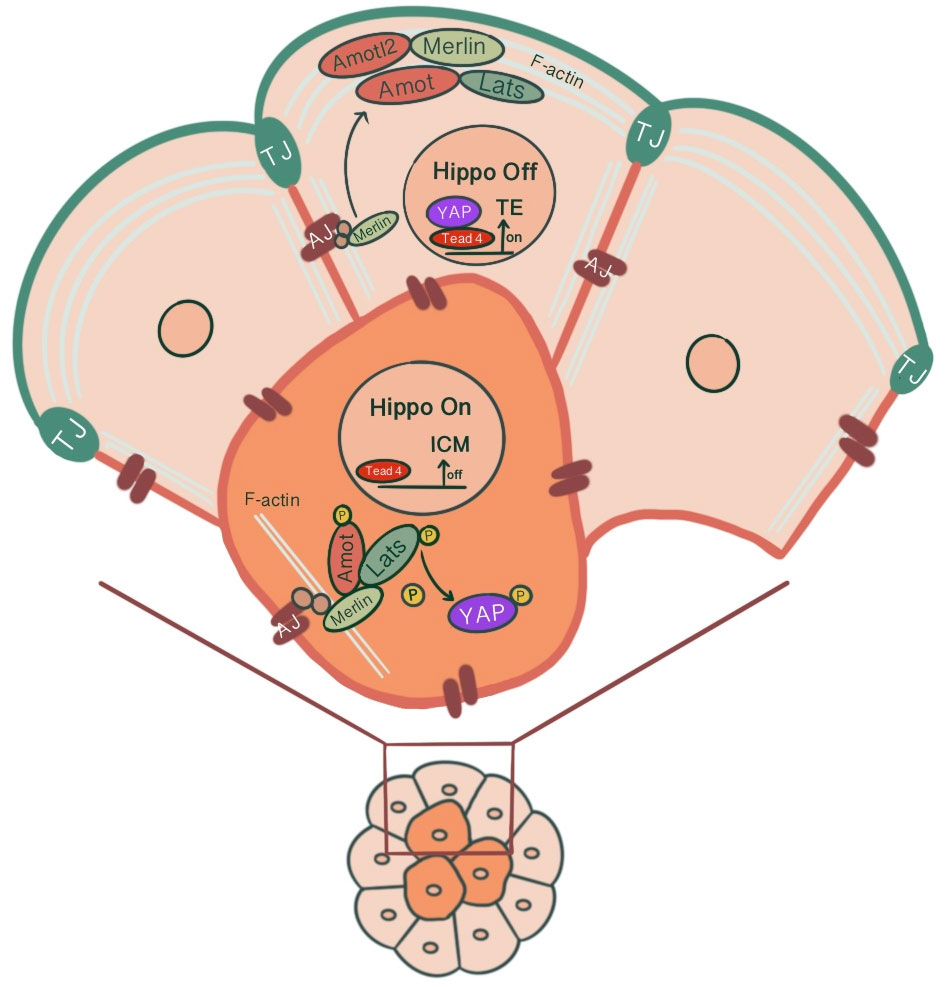
2.1.3 Cell fate determination by differential activation of the Hippo signaling pathway in inner and outer cells through E-cadherin & apical polarity complex association with AMOTThe transition from inner/outer cells to pluripotent ICM/TE lineages is achieved through the control of the Hippo signaling pathway and its effects on lineage-specific gene expressions (46). For TE specification, Caudal-related homeobox 2 transcription factor (Cdx2) expression along with POU-family transcription factor Oct3/4 and Nanog repression is required. Cdx2 expression that starts ubiquitously at the time of polarization progressively increases in outside cells, eventually leading to positive regulation of trophectoderm lineage markers (47). Pluripotent embryonic stem cells show morphological differentiation to TE lineage by overexpression of Cdx2 and similarly by forced repression of ICM-specific transcription Oct4 (48, 49). The differential activation of target genes via Hippo signaling in outer/inner cells is achieved by modulation of the activity of transcription factor Tead4 through the differential localization of Tead4 coactivator protein Yap. The inner cells, with the activation of the Hippo pathway, Yap phosphorylation by Lats kinase1/2 sequesters Yap in the cytoplasm, causing Tead4 to remain inactive, repressing target gene expression. However, in the absence of Yap phosphorylation, Yap’s nuclear accumulation and Tead4 activation induce Cdx2 expression in outer cells, promoting TE fate (27, 50) (Figure 3). As a result, a blastocyst with two different cell populations is formed.
Figure 3 Differential modulation of the Hippo signaling pathway in inner and outer cells of the preimplantation embryo, influenced by junction-associated scaffold protein angiomotin (Amot) and its interaction with tight junctions & adherens junctions. In outer cells, Amot co-localizing with ZO-1 causes apical F-actin-mediated suppression of the Hippo signaling pathway. Through sequestration of Amot to the apical membrane domain via the Par-aPKC complex, Yap, which can translocate to the nucleus, causes outer cells to adopt TE fate via Tead4 activation and Cdx2 expression. In inner cells, the E-cadherin-β-catenin-α-catenin-Merlin-Amot complex acts as an upstream regulator of the Hippo signaling pathway through Yap sequestration at cytoplasm, repressing target gene expression.
Intercellular adhesions play a critical role in determining cell fate and establishing a position dependent Hippo signaling through their effect on cell polarity. (7). In E-cadherin mutant embryos, although a blastocoel cavity or TE do not develop, differential expression of Cdx2 and Oct4 is still observed. However, it occurs in a disrupted ratio, with more cells expressing Cdx2 in mutant embryos compared to the wild-type. Cdx2(-/-) embryos fail to maintain blastocoel and epithelial integrity due to disrupted localization of ZO-1 α-, ZO-1 α+ and E-cadherin (51). Thus, although individual cells can initiate ICM- or TE-like fates, E-cadherin is necessary for correctly allocating cell fate and normal spatial distribution of ICM- and TE-like cells (30). Bovine zygotes treated with E-cadherin dsRNA demonstrate significantly lower blastocyst formation rate (52). E-cadherin has a unique function in TE formation. Gene replacement by N-cadherin cDNA introduction into the E-cadherin genomic locus and inactivation of maternal E-cadherin allows compaction and expression of epithelial makers in outer cells. However, a fully polarized epithelium, TE, cannot form due to the failure to correctly assemble junctions and cytocortical networks (53).
It has been suggested that junction-associated scaffold protein angiomotin (Amot) acts as a molecular switch for Hippo signaling pathway. In inner cells, Amot causes activation of Hippo signaling through its interactions with AJs. In contrast, in outer cells, there is an apical F-actin mediated suppression of the Hippo signaling pathway (54). In 16 cell stage, Amot becomes differentially localized among inner/outer cells. While it is localized to the apical membranes of outer cells, inner cells demonstrate a distribution throughout the membranes. This is maintained until the early blastocyst stage. In non-polar inner cells, Amot localizes at AJs through interacting with Merlin by its coiled-coil domain, forming a large complex of E-cadherin-β-catenin-α-catenin-Merlin-Amot, which acts as an upstream regulator of the Hippo signaling pathway. N-terminal of Amot mediates actin binding, Nf2/Merlin mediated interactions with E-cadherin and associations with Lats kinases (55). Phosphorylation of Amot by LATS1/2 kinases suppresses the actin-binding activity of Amot and causes its mislocalization from cortical F-actin in the junctional sites stabilizing it in the basolateral AJs. This adds another layer of regulation to the Hippo signaling pathway where phosphorylation of Amot acts as a switch for phosphorylation and cytoplasmic sequestration of Yap (55–57). Maternal-zygotic Nf2 mutant embryos show defects in Yap localization and Cdx2 expression, where inner cells resemble TE and ultimately lose their normal ICM identity (58). In MDA-MB-231 cell line, it has been shown that doxycycline-induced expression of E-cadherin and homophilic binding of E-cadherin between two adjacent cells cause redistribution of Yap from the nucleus to cytoplasm. Additionally, endogenous depletion of β-catenin in MDA-MB-231 cells cause increased nuclear accumulation of Yap and phosphorylation on the S127 residue (57). Similarly, cell contact inhibition using E-cadherin blocking antibody ECCD1 inhibits the nuclear accumulation of Yap in the inside cells of the morula (50). Correspondingly, Yap knockdown in porcine embryos significantly reduced development to 8-cell and blastocyst stage, TE cell number and increased Cdx2 negative cells. Additionally, decreased gene expression involved with cell fate specification (Cdx2, Tead4, Oct4, Sox2, Nanog), junction assembly (Ocln, Cldn4, Cldn6, Cldn7, Tjp1, Tjp2, Cdh1), and fluid accumulation was evident (59) (Figure 3). A study showed that knockdown of AP-2γ (Tcfap2c), a novel upstream regulator of Cdx2 expression, causes downregulation of Pard6b, Tjp2, Cldn4, Cldn6, Cldn7 and prevents TJ formation and paracellular sealing at apical cell borders, inhibiting blastocyst formation (60, 61). Knockdown of INO80 in porcine embryos impaired blastocyst development, paracellular sealing of TE, and decreased expression of OCT4, CDX2, TEAD4, CDH1, OCLN and several cell polarity, cytoskeleton and fluid-accumulation related genes (62).
In the outer cells, Amot is present at TJs, co-localizing with ZO-1. Par-aPKC complex sequesters Amot to the apical membrane domain, held in an inactive state by interacting with F-actin (54, 55). As a result, unphosphorylated Yap can enter the nucleus. Pard6b, a homolog of the Par-6 gene, knockdown causes failure in blastocoel cavity formation due to defective TJ formation, abnormal cell polarization, abnormal distribution of actin filaments and, as a result, diminished expression of Cdx2 (63) (Figure 3).
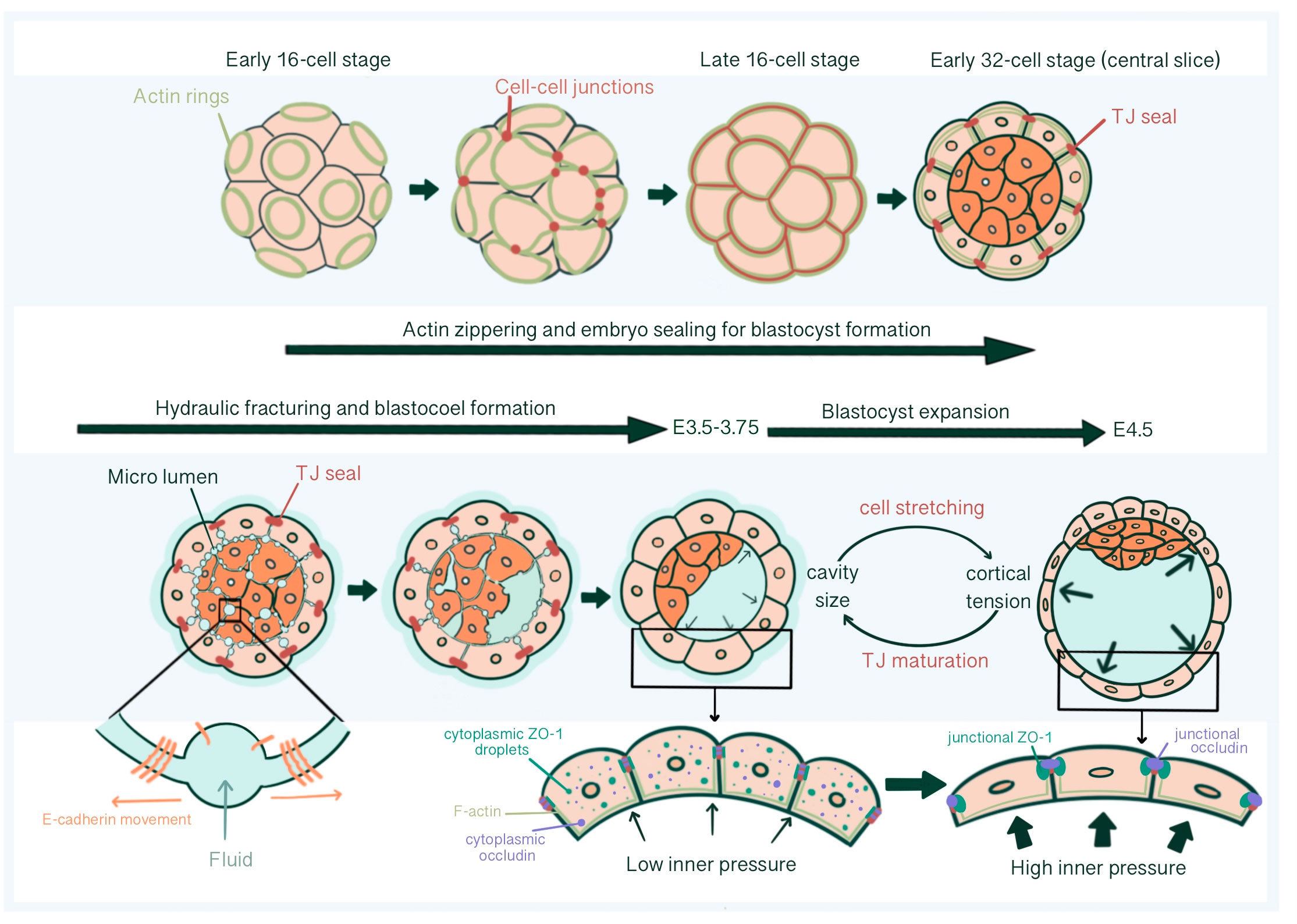
2.1.4 Directing hydraulic fracturing during blastocoel formation by reorganization of E-cadherin at cell-cell contactsBlastocoel cavity formation starts as micro-lumens through a process of hydraulic fracturing. Micro-lumens form at cell-cell contacts causing fluid accumulation from the outside environment into intercellular space regarding an osmotic gradient, established by differential Na+ concentration by Na+/K+ ATPases at the basolateral membrane together with basolateral Aquaporin 3 and 8 mediated trans-cellular water influx (64–67). For this, E-cadherin must be redistributed and accumulate at micro-lumen edges, causing a separation of previously cohesed cell membranes (65). The intercellular connections of micro-lumens allow fluid to move from smaller micro-lumens into larger ones, parallel with the Young-Laplace equation, creating one large, dominant micro-lumen: the blastocoel (66) (Figure 4).
Figure 4 Tight junction seal formation and force dependent junctional maturation, enabling hydraulic fracturing and blastocoel expansion, from E2.75 until E4.5. The actin ring structure formed in the apical region of the blastomeres in the early 16-cell stage embryos affects intercellular junction maturation and formation of TJ sealing. With hydraulic fracturing coordinated via reorganization of E-cadherin at cell-cell contacts, fluid accumulates within the embryo forming the blastocoel The integrity of blastocyst depends on mature TJ seals that prevents collapse with increasing inner pressure. Force dependent (increased luminal pressure and cortical tension, accompanied with TE cell stretching) junctional maturation enables accommodation to pressure increase and blastocoel expansion prior to hatching.
2.2 β-cateninβ-catenin has a dual function as a transcriptional effector of Wnt signaling and a constituent of AJs (68). Wnt signaling is an important regulator of the maintenance, self-renewal and differentiation of adult mammalian tissue stem cells (69–71). In addition, it has been demonstrated to regulate major embryonic events such as anterior-posterior patterning (72) transcriptional programmes at gastrulation (73) specification of the primitive streak and distal visceral endoderm (68, 64, 75). In the canonical Wnt pathway, β-catenin functions as a transcriptional co-activator. In the absence of Wnt, β-catenin undergoes proteasomal degradation by a destruction complex residing in the cytoplasm, preventing it from reaching the nucleus and interacting with Wnt target genes. When Wnt binds to a heterodimeric receptor complex, consisting of Frizzled receptor and its co-receptor LRP6 or LRP5, Wnt-Fz-LRP6 leads to inhibition of Axin-mediated β-catenin phosphorylation. β-catenin is stabilized and translocated into the nucleus, forming complexes with TCF/LEF and activating Wnt target gene expression (76). Transcripts of ligands and antagonists of the Wnt signaling pathway were detected in the mouse preimplantation embryos. Wnt3a and Wnt -4 start to be detected in 4-cell and precompact 8-cell stage embryos, with robust enhancement in expression with compaction. Wnt3a transcripts in 2-cell stage embryos have not been specifically determined whether they are from a maternal or embryonic origin (77). Wnt-1, -3, -3a, -4, -5a, -5b, -6, -7a, -7b, -9a, -10b, -11, -13, and Wnt signaling antagonists Sfrp1 and Dkk1 are all present in mouse blastocysts, detected by qRT-PCR (78–82). Amongst, Wnt-3a, -6, -7b, -9a and -10b have the highest gene expression levels (81, 82). With exposure to uterine factors such as estradiol surge during the morula to blastocyst transition, Wnt-11 demonstrates upregulation during in vitro blastocyst development (78). In addition, the expression patterns of Wnt ligands differ throughout the blastocyst, with Wnt-1 predominantly expressed in ICM, Wnt-3a, -6, -7b and -10b in the whole blastocyst, and Wnt-9a in mural trophoblast and cells that surround the forming blastocoel cavity, demonstrated with whole-mount in situ hybridization (81, 82). Single-cell RNA-seq profiling of late human blastocysts demonstrate upregulation of the genes encoding the Wnt signaling pathway receptors in epiblast cells (EPI) and primary outgrowth during human embryonic stem cell derivation (83).
Maternal/zygotic Ctnnb1 null blastocysts undergo normal first and second lineage specification. However, the blastocysts are small and undergo fission after hatching or removing the zona pellucida. Fission results in the formation of trophoblastic vesicles that can undergo decidual reactions but not sustain further embryogenesis. Therefore, Messerschmidt et al. suggest that β-catenin-mediated adhesion is important to maintain adhesion between blastomeres at the compaction and blastocyst stage to enable subsequent embryonic development and prevent fragmentation (84).
Wnt signaling is necessary for the allocation of ICM and TE lineages in the blastocyst, regulating hatching, and ensuring blastocyst competency for implantation (21, 85–87). Several studies found that activation of Wnt signaling was inversely correlated with the ability of blastocyst hatching. For example, pig blastocysts treated with Dkk1 demonstrate an increased ability to hatching on day 7 and day 8 of pregnancy (85). Similarly, in mouse blastocysts, Wnt activation by LiCl significantly decreased blastocyst hatching rate in a dose-dependent manner, in addition to decreased subsequent adhesion and outgrowth on fibronectin (22). In bovine blastocysts, activation of the canonical Wnt pathway by Wnt agonist AMBMP caused reduced development to the blastocyst stage (88). In contrast, in a different study, exogenous activation of canonical Wnt pathway via 6-Bio treatment enhanced bovine blastocyst development and hatching rate, with significantly increased Oct4 in ICM and decreased Cdx2 expression within the ICM and TE compared to the control (89). This suggests that the Wnt pathway positively affects the maintenance of pluripotency marker genes synergistically with PPARδ expression during first lineage diversification (89, 90). These results are in line with previous reports in pig blastocysts where Wnt activation through LiCl treatment causes a lower TE/ICM ratio due to a reduced number of total blastomeres and TE cells (85), in bovine embryos where Wnt inhibition by Dkk1 exposure during morula to blastocyst transition causes increased differentiation into TE and hypoblast lineage (86), and in ICM-derived embryonic stem cells where Wnt activation provides maintenance of pluripotent state and undifferentiated phenotype (91–93). In another study, the inactivation of nuclear β-catenin signaling in mouse blastocysts did not affect the development to the blastocyst stage. However, blockage of nuclear β-catenin accumulation significantly reduced Cdx2 expression in TE and impaired normal implantation, potentially by downregulating RhoA GTPase, causing disassembly of AJs and cytoskeletal reorganization (21). Likewise, in human embryos treated with β-catenin degrading drug Cardamonin, blastocysts development rate, CDH1, Nanog, and Sox2 expression levels were unaffected. However, significantly fewer TE cells with Cdx2 positivity supported β-catenins’ role in TE lineage specification (23). The downregulation of β-catenin not interfering with development until the blastocyst stage can possibly be explained by the ability of plakoglobin to rescue the function of β-catenin (23, 94).
With exogenous Wnt stimulation, there is enhanced spatial overlapping of PPARδ and β-catenin under immunofluorescence analysis. Similar to the effects of canonical Wnt/β-catenin inhibition, blocking of PPARδ in bovine blastocysts causes significantly reduced cell proliferation ratio, blastocyst quality, cell invading ability, and weak blastocyst attachment (89). For instance, overexpression of Dkk1 blocks the activation of dormant blastocysts for implantation in response to E2 injection on day 7. However, exposure to Wnt-3a partially overrides this effect via nuclear β-catenin stabilization and PPARδ expression as 11% of Wnt-3a treated blastocysts gain implantation competency, highlighting the necessity of nuclear β-catenin signaling in blastocyst activation for implantation (21). In addition to the synergistic effects of PPARδ and β-catenin on proliferation and lineage specification, the coordination of Wnt-β-catenin with PPARδ signaling regulates lipid metabolism, enhancing the blastocyst development and implantation potential (21, 89, 90). Lipid metabolism and ATP generated through fatty acid oxidation is an important energy source for early embryonic development (95, 96). When Wnt stimulation induces PPARδ activity, there is a significant reduction in lipid droplet content, indicative of a high fatty acid oxidation metabolism (89).
Furthermore, Wnt signaling is indispensable for early placentation, trophoblast invasion and differentiation (97). Wnt-3a induces cytoplasmic accumulation of active β-catenin and translocation into the nuclei of differentiating trophoblast stem cells causing upregulation of c-Myc and PPARδ, establishing Wnt-β-catenin signaling as a regulator of trophoblast differentiation (21, 90). High level of β-catenin accumulation in the TE cells is correlated with an increase in cell migration and invasion capacity (90). Similarly, in human embryos exposed to Wnt-3, trophoblast specific marker EOMES is upregulated, supporting the necessity of Wnt signaling for TE specification to the trophoblast lineage (23). More studies with genetic manipulations of PPARδ will help enlighten the coordination between Wnt and PPARδ during early embryonic development.
Haegel et al. showed that embryonic ectoderm and mesoderm development is significantly disrupted in β-catenin null mutant mouse embryos (75). In mouse embryos derived from oocytes expressing a stabilized form of β-catenin resistant to GSK3β-mediated proteosomal degradation, development to the blastocyst stage is morphologically normal. However, mutant embryos exhibit a distinct phenotype at E6.5, with a less expanded and disorganized embryonic portion. Cells of the embryonic ectoderm in early postimplantation embryos change their fate, leading to premature epithelial-mesenchymal transition and loss of E-cadherin transcription, supporting the necessity of β-catenin in developing tissues derived from ICM and the maintenance of differentiation potential (98). Therefore, Wnt-β-catenin signaling also contributes to proximo-distal patterning, possibly by generating a symmetry-breaking signal and distinct cell-specification in ICM (99).
3 DesmosomesDesmosomes are adhesive intercellular junctions that tether intermediate filaments (IF) and provide mechanical stability and tissue integrity against mechanical stress (100). Although AJs and desmosomes both confer adhesive properties to tissues, three factors set desmosomes apart from AJs: IF’s ability to stretch to multiple times its original length, IF’s capacity to withstand higher tensile loads than actin and desmosomes’ potential to transform into calcium-independent “hyper-adhesive state” (101). Ultrastructurally, desmosomes are disc shape electron-dense plaques which are 0.2-0.5 μm in diameter. They are composed of a 20 nm thick dense outer plaque, a 7 nm thick dense inner plaque where IF makes a loop attachment and a 34 nm intercellular space with a discrete electron-dense midline creating a mirror image arrangement at cell contact sites (102). Prominent midlines are a characteristic of hyper-adhesive desmosomes, whereas Ca-dependent desmosomes exhibit a somewhat amorphous intercellular space (103).
Desmosomes comprise a membrane core of desmosomal cadherins, desmogleins and desmocollins; a cytoplasmic plaque of armadillo proteins, plakoglobin and plakophilins; and a cytoskeletal adaptor that mediates IF anchorage, desmoplakin (101, 102, 104) (Figure 1). Desmosomal cadherins, which constitute desmogleins and democollins, are transmembrane glycoproteins that comprise four extracellular cadherin homology domains, each separated by calcium-binding motifs, a fifth extracellular anchor domain, a single transmembrane domain and a cytoplasmic domain consisting of intracellular anchor and a cadherin-like sequence (ICS). Desmogleins additionally pose unique motifs like a proline-rich linker domain (IPL), a repeat unit domain (RUD) and a desmoglein terminal domain (DTD) (104). There are three isoforms of desmocollin (Dsc1-3) and four isoforms of desmoglein (Dsg1-4) in humans (102). Each of these three isoforms of desmocollins can be alternatively spliced to generate longer “a” and shorter “b” forms that differ in their ICS domain length. Plakoglobins (PK), β-catenin orthologue, and plakophillins (PKP), member of the p120-catenin subfamily, act as a bridge between the ICS domain of desmosomal cadherins and desmoplakin through their central armadillo domain. Structurally, in addition to amino- and carboxyl-terminal domains, PK contains 12 arm repeats, and PKP contains 9 arm repeats with an insert between the fifth and sixth repeats that creates a bend in its morphology (104). Plakin family member desmoplakin, the adapter that couples IF to the desmosomal plaque, contains globular amino and carboxyl terminals and a central a-helical coiled-coil rod domain. Glycine-serine-arginine rich domain (GSR) of desmoplakin carboxyl-terminal mediates the binding of IF (102, 104).
Desmosomes first assemble at cell-cell contact points of TE in the early blastocyst (32-cell stage), in close relation to the onset of blastocyst cavitation and increase in number as the blastocoel expands (105, 106). Immunoprecipitation studies show that desmoplakins and desmogleins are absent in unfertilized eggs. The earliest detectable desmosomal constituent is plakoglobin at compaction (72 h post hCG injection) followed by desmoplakin 1 and 2 at the 16-cell stage (84 h post hCG). Finally, desmogleins and desmocollins start to be detected at the early blastocysts (96-100h post hCG) (105).
Immunocytochemistry studies reveal that desmoplakin (DP) 1&2 first appears as faint punctuate stains along with lateral membrane contact sites after division into 32-cell stage, 2-4h hours post-cavitation, localized exclusively to outer polar TE cells. DP staining becomes stronger in later blastocysts, 12-48h post cavitation. It may be argued that as DP expression in the TE is concomitant with blastocoel cavity formation, DP might be essential in mechanical stability against stress during blastocoel cavity formation (105). However, homozygous DP(-/-) mutants form a TE layer, blastocoel cavity and proceed through implantation (107). Nevertheless, during post-implantation, embryos die by E6.0-E6.5 because of defects in extra-embryonic tissues due to disruption of the keratin network and a dramatic reduction in the number of desmosomal-like junctions at endoderm and ectoplacental cone (106, 107). When Dp(-/-) embryos are rescued by supplementing with Dp(+/+) extra-embryonic tissues, they die shortly after gastrulation, approximately at E12.5, due to overall heart architecture defects, malformed neuroepithelium, collapsed neural tube, fragile skin epithelium and significantly reduced and disrupted capillaries (106). Therefore, DP is essential in early embryonic development through anchoring and maintaining keratin intermediate filament network and assembling/stabilizing desmosomes, all of which are at the very least critical during egg cylinder formation and development of surface ectoderm that can withstand mechanical stress enabling post-implantation growth (107).
Plakoglobin (PG) mRNA is detected very faintly at unfertilized eggs and 2-cell stage embryos and increases from 8-cell stage onwards. Immunoblot studies demonstrate very low abundant PG protein levels from unfertilized egg until early morula stage, with an increase from late morula satge onwards (14). PG are present in the majority of late uncavitated morula under the immunofluorescence microscope as faint linear staining at borders between outer cells. In cells clusters derived from 2/16 couplets, PG linear membrane localization exhibits a similar pattern to DP 1&2, occurring after division into a 32-cell stage (105). However, while 2/16 cell cluster studies reveal that DP 1&2 staining only occurs at presumptive TE cells of cavitated embryos filled with fluid, PG is detected both in cavitated and non-cavitated cell clusters indicating its independence from blastocoel formation. PG also demonstrates a different spatial regulation than DPs. At the onset of PG labeling in cell clusters, PG appears in all cell-cell contact points, including ICM, in probably a non-desmosomal localization associated with vinculin or α-actinin. Later on, PG becomes localized to TE in late blastocysts, like other desmosomal proteins (105). Although the inactivation of plakoglobin gene does not affect basic morphogenetic events until early post-implantation, PG mutant embryos die from E10.5 onwards due to severe heart defects, especially in ventricular and atrial trabeculae and endocardial cushions (108).
Desmogleins (DSG) are first detectable as faint punctate stains under immunofluorescence microscopy 2-4 h post-cavitation in early blastocysts and become prominent in TE lateral membranes of expanded blastocysts 12-48 hours post-cavitation (105). Germline inactivation experiments of DSG2 indicate that DSG2 function is essential for embryonic viability at the time of implantation. All DSG2(-/-) mice and a considerable number of DSG2(+/-) mice show a lack of decidual reaction and die at or shortly after implantation (109). Loss of DSG2 also affects the distribution of DP, disturbing its normal localization at cell borders of the blastocyst (109).
Desmocollin 2 mRNA (DSC2) is detected in cumulus cells, unfertilized eggs, 2-, 4-, pre-compact and 16-cell stages with the exclusion of compact 8-cell stage, coinciding with maternal DNA degradation, indicating the contribution of both maternal and embryonic genomes in desmocollin expression (110). DSC3 mRNA is present in unfertilized eggs as well as pre-compaction 8-cell stage embryos (111). Both a and b isoforms of DSC2 and DSC3 are transcribed in the pre-implantation embryo (110, 111). DSC2 transcription through the embryonic genome is initiated at 16-cell and 32-cell stages just before DSC protein starts to become detectable by immunoprecipitation (110). The earliest detectable linear expression of DSC2 and 3 is present in expanding blastocysts, 12-48 h post cavitation, confined to TE cells and becomes punctate in late blastocysts. So, the maturation pattern of desmosomal proteins under immunofluorescence labelling appears as a change from linear to punctate distribution, restricted to TE cells, with exclusion of PK (105, 111). However, DSC3 might not be restricted to classical desmosomes of early developmental stage embryos. Den et al. showed a similar staining pattern for DSC3 to E-cadherin and β-catenin: linear staining along cell-cell borders of TE cells. This staining pattern may indicate DSC3’s role in maintaining cell-cell adhesion and mechanical integrity of early cleavage-stage embryos alongside E-cadherin. DSC3 staining is also observable in the cytoplasm of oocytes with immunohistochemical staining of ovary sections, indicating that DSC3 is not specifically distributed to desmosome-like cell junctions (111). Dsc3(-/-) homozygous mutant embryos die before compaction is completed, most disintegrating within E2.5 (111).
There is a close relationship between cavitation and desmosome biogenesis, so cavitation might be a prerequisite for triggering desmosomal cadherin synthesis. Correspondingly, desmosomes may be essential in stabilizing TE against fluid pressure within the blastocoel during blastocyst expansion (105). As DSG and DSC expression supersedes the synthesis of desmosomal plaque proteins and coincides with desmosome assembly in TE during cavitation, Collins et al. proposed that DSC2 expression regulates desmosome biogenesis (110).
Intermediate-sized (7-11 nm) filaments produced during pre-implantation embryo development are bundles of cytokeratin filaments and are present in the outer cells of morula and the trophectoderm in close association with desmosomal structures (112). Cytokeratin filament polymerization requires one acidic cytokeratin, ENDO B, and one neutral/basic cytokeratin, ENDO A (113). Synthesis of ENDO A and ENDO B is detected in low levels at the 4-to-8 cell stage by immunoprecipitation (114). mRNA of ENDO A was detected in 8-cell embryos; however, 4-cell embryos were not probed directly (115). Cytokeratin filaments initiate to assemble in a cell-autonomous manner influenced by differential contact patterns. There is an extensive filament network formation in the outer cells compared to relatively low levels in the inside cells (113). In the late morula stage embryo, intermediate-sized filaments are related to small areas of nascent desmosomes. In the blastocyst, tonofilament bundles are anchored to normal-sized desmosomes with typical architecture. Just like desmosome-like junctions, intermediate filament structures are not found in ICM (112).
De novo keratin network biogenesis has been demonstrated in homozygous keratin 8-YFP knock in mice, that produces fluorescence-tagged keratin 8. 6-8 hours after compaction, diffuse cytoplasmic signals appear and increase continuously at cell borders with maturation into the blastocyst stage, perfectly co-localizing with DP and immediately next to DSG2 (116, 117). The punctate accumulation in TE of the early blastocyst evolves to a pearl-on-a-string model consisting of elongated puncta connected by subcortical filaments in the late blastocysts. Moch et al. suggest that nascent desmosomes serve as nucleation sites for elongating keratin filaments. With subsequent desmosomal fusion, keratin filament anchorage, elongation and bundling, the keratin-desmosome connectivity is stabilized (117).
Desmosomes of the trophectoderm become hyper-adhesive in the blastocyst stage, proceeding from E3 to E4.5. In most early blastocysts, DP is internalized under low-calcium medium incubation (118). However, late blastocysts show DP staining on their plasma membranes, with no sign of DP internalization under low-calcium medium incubation, indicating the desmosome’s transformation into a Ca-independent state. The acquisition of hyper-adhesiveness is correlated with junctional maturation. While Ca-dependent desmosomes of early blastocysts are ultrastructurally rudimentary, without a distinct midline, little IF attached to poorly developed cytoplasmic plaques; Ca-independent hyper-adhesive desmosomes of late blastocysts are mature junctions with well-developed cytoplasmic plaques and abundant IF and a prominent electron-dense midline (118).
Desmosomal re-arrangement between uterus and TE is one of the various factors contributing to successful implantation into the endometrium. In murine maternal luminal epithelium, desmosomal adhesions between trophectoderm cells are downregulated and re-arranged between uterus and trophectoderm, facilitating endometrial invasion (119). Desmoplakin protein expression progressively decreases during the pre-implantation period of uterinal epithelium and becomes barely detectable by days 4.5-5 of pregnancy (120). After hatching, initially hyper-adhesive desmosomes need to weaken during migration of trophoblast cells and revert to calcium dependence. When desmosomal adhesiveness was evaluated in E4.5 blastocysts for 96 h in a low calcium medium, during the time which extensive migration of the TE occurs, internalization of DP molecules was demonstrated. In contrast, blastocysts in a standard medium demonstrated desmosomal junctions in cell-cell contact sites. These changes under a low calcium medium indicate a migratory phenotype where desmosomes are reverted to a calcium-dependent state to enable trophectoderm invasion and successful implantation of the embryo (118).
4 Tight junctionsTJs, also named zonulae occludens, are the most apical structures of the apical junctional complex, followed by AJs and desmosomes (121). They seal the paracellular space creating a near leak-proof permeability barrier. TJ have two main functions: fence and gate function (5). As the fence function, TJ provides membrane polarity by separating the plasma membrane into apical and basolateral domains, and creating an asymmetry regarding the composition of cytosol and plasma membrane proteins and lipids. As the gate function, TJ establishes a paracellular diffusion barrier between sealed cells and regulates the passage of solutes and ion selectivity. The structure of TJ greatly affects the transepithelial junctional resistance, thus the transepithelial permeability characteristics (122). TJ barrier function contributes to the structural integrity of cellular sheets and the prevention of invasion of pathogens. Moreover, TJ regulates cytoskeletal protein organization, controlling the actomyosin contractility and distributing cytoskeletal generated tensional forces (6).
TJ comprises three main transmembrane proteins: junctional adhesion molecules (JAM), claudin and occludin. Scaffolding proteins such as zonula occludens proteins (ZO-1, ZO-2, ZO-3) and cingulin associate occludin, claudin and JAM in tight junctional strands, promoting polymerization. These cytosolic plaque proteins also integrate inside and outside signaling and enable binding with cytoskeletal-associated proteins (123) (Figure 1).
During embryonic development, TJs have critical roles in several morphogenetic events: 1) Generating apicobasal polarity within blastomeres. 2) Mediating differential AMOT localization at apical membranes of outer cells affecting the cell fate decisions. 3) Creating a functional barrier that triggers cavitation and formation of a fluid-filled blastocoel through hydraulic fracturing and basolateral Na+/K+ ATPase mediated fluid expansion. 4) Maintaining blastocyst integrity through paracellular sealing (65, 67, 124–125). Apicobasal polarity established in earlier stages of development has two important roles subsequently during blastocoel cavity formation in the blastocyst stage: 1) generation of transepithelial transport by basolateral Na+/K+ ATPase via establishment of an electrochemical gradient and osmotic flow of water and 2) apical TJ biogenesis forming a permeability seal that prevents blastocoel collapse (67, 126). Role of individual TJ proteins regarding these specific events throughout the embryo development will be explained separately.
4.1 ClaudinsClaudin family is composed of four transmembrane domains, two extracellular loops, a short cytoplasmic turn, and an amino- and carboxyl-terminal containing a PDZ binding motif, which binds to PDZ domain-containing plaque proteins such as ZO (127). Claudin is the major structural component of TJs, forming the backbone of TJ strands, whereas other proteins regulate TJ dynamics (128). It is the primary regulator in defining TJ functional properties, such as creating a tight paracellular cleft and regulating paracellular permeation. Therefore, the claudin family members are subdivided into sealing and channel-forming proteins (6). It has been shown that overexpression of claudin-1 in MDCK cells causes decreased paracellular flux and increased transepithelial electrical resistance, supporting its role in epithelial barrier function (129). Claudin’s sealing role is supported in various claudin knockout experiments, as Cldn1(-/-) mice die within a day after birth as a result of excessive dehydration, Cldn5(-/-) mice show a severely disturbed blood-brain-barrier and die within a day of birth and Cldn 11(-/-) male mice show infertility due to disruption of blood-testis barrier and spermatogenesis (130). Claudin can associate with enzymes such as protein kinases and matrix metalloproteinases (38) and are post-translationally regulated on their CxxC motifs by palmitoylation and phosphorylation (127).
Out of 24 members of the claudin gene family, only Cldn 4, 6, 7 and 12 are detected with RT-PCR starting from the pre-compact 8-cell stage of the mouse embryo. In double immunofluorescence staining with occludin; claudin 4, 6 and 7 were detected as punctate stains in some of the compact 8-cell stage embryos in cell-cell contact regions and converted into continuous bands in every 16-cell stage embryo. In blastocyst, claudin and occludin are co-localized at the most apical regions of the cell interface in TE. To evaluate the contribution of claudins to TJ seal and blastocyst formation, C- terminal of Clostridium perfingens enterotoxin (C-CPE) was introduced into culture medium to bind with extracellular domains of claudin 4 and 6. The barrier function of C-CPE treated mouse embryos was significantly disrupted, interpreted by infiltration of FITC-dextran, a 4kDa permeability tracer, into the blastocoel cavity at E4.0 (131). Ordinarily, the permeability seal should have been established around E3.5. Morphologically, blastocoel cavitation was mostly immature, and in those with a definite blastocoel, expansion was inhibited. Furthermore, C-CPE treatment caused a developmental delay of the mouse embryos, as Oct 3/4, a pluripotency marker, was expressed in all cells at E4.0, which should have been excluded to ICM by E4.0. However, final differentiation was not affected as Cdx2 was expressed in TE and Oct 3/4 at ICM, thus establishing cell fate (131). Similar effects were seen in the knockdown of Cldn7 in porcine embryos. Cldn7 knockdown reduced the expression of genes related to TJs (Ocln, Cldn4, Cldn6, Cdh1, Tjp1, Tjp2) and several others associated with cell polarity, cytoskeleton and H2O channels. Consequently, TJ paracellular sealing was disturbed, as higher FITC-dextran permeability was shown in Cldn7 knockdown embryos. Furthermore, developmental competence was reduced, but no influence on Sox2 or Cdx2 expression was detected (132).
In single-cell RNA-Seq profiling of human preimplantation embryos, the differential expression pattern of Cldn10 and Cldn3 is demonstrated between EPI and TE cell lineages of the cells in late blastocysts, providing insight for understanding gene regulatory networks of embryonic development (33). Similarly, transcriptome analysis of human TE cells demonstrated 2,196 transcripts specific to TE molecular signature, including genes related to junctional proteins such as Ocln, Dsc2, Dsp, Jup, Pkp4, Gja5, and Vcl. Amongst, Cldn4 was one of the 100 genes with highest fold change and significant statistical value in TE samples (133). Hernandez-Vargas et al. identified 24 common potential biomarkers related to reproductive outcomes by examining all available endometrial and embryonic omic studies, where Cldn4 is present for its role in the interaction between the embryo and uterus demonstrated with interactome analysis. Through functional enrichment analysis of embryonic and endometrial interaction networks, ZO-1, occludin and claudin 4 were found to be involved in one of the largest interaction networks at the time of implantation. Therefore, it is important to emphasize the place of claudin within embryonic and endometrial molecular profiles and its contribution to successful preimplantation development and implantation (134, 135).
4.2 Junctional adhesion molecules (JAMs)The IgG-like family of JAMs is composed of single-pass membrane protein, forming homophilic and heterophilic interactions with intrafamily and extrafamily partners and constitute two immunoglobulin-like domains, one transmembrane domain, one cytoplasmic tail containing a PDZ domain, and a zonula occludens protein-binding motif (136). This family consists of three classical (JAM-A, JA
留言 (0)