
記住我
This was an open-label, multiple-dose, randomized, parallel-arm, multicenter trial in adults with schizophrenia or BP-I (ClinicalTrials.gov identifier: NCT04030143). The study started on 1 August 2019 and was completed on 8 July 2020. Participants were enrolled into the study at 16 clinical trial sites in the United States (US).
Key inclusion criteria were: age 18–64 years, current diagnosis of schizophrenia or BP-I (as defined by Diagnostic and Statistical Manual of Mental Disorders, 5th edition [DSM-5] criteria) [1], body mass index of 18–35 kg/m2, good physical health, clinical stability on an atypical antipsychotic medication (except for clozapine, which was not allowed) for ≥2 months prior to screening (participants with BP-I were also allowed to continue concomitant mood stabilizer and antidepressant treatment), and prior history of tolerating oral aripiprazole and/or AOM 400 (according to the investigator’s judgment). Participants without a history of tolerating aripiprazole received three single oral aripiprazole doses of 10 mg on 3 consecutive days (30 mg in total, in addition to their current oral antipsychotic) during the screening period to establish tolerability.
Key exclusion criteria were substance use disorder (as defined by DSM-5 criteria) [1] within the past 180 days or a positive test for drugs of abuse (excluding nicotine, alcohol, and marijuana if sufficient rationale was provided, which must have included rationale/indication for use, review of patterns and frequency of use, and justification to support compliance with the protocol for the intended duration of treatment); use of any cytochrome P450 (CYP) 2D6 and CYP3A4 inhibitors or CYP3A4 inducers within 14 days (fluoxetine or fluoxetine/olanzapine within 28 days) prior to dosing, for the duration of the trial, and 30 days after the last study drug dose; current acute relapse of schizophrenia; current DSM-5 diagnosis other than schizophrenia or BP-I [1]; a significant risk of committing suicide (based on history, routine psychiatric status examination, investigator’s judgment, or a ‘yes’ answer to questions 4 or 5 [current or over the last 6 months] on the baseline Columbia Suicide Severity Rating Scale [C-SSRS] questionnaire); treatment resistance to an atypical antipsychotic medication; and history of neuroleptic malignant syndrome or clinically significant tardive dyskinesia (as assessed by the investigator).
The study design is shown in Fig. 1.
Fig. 1
Study design. a Participants received overlapping oral antipsychotic treatment with the first study drug dose to ensure antipsychotic treatment continuity. No overlapping oral antipsychotic treatment was administered to participants stabilized on AOM 400; these participants could not be enrolled to the robust sampling schedule. AOM 400 aripiprazole once-monthly 400 mg, Ari 2MRTU 960 aripiprazole 2-month ready-to-use 960 mg, BP-I bipolar I disorder, ET early termination, R randomization
Participants were randomized (1:1) to the two treatment arms (Ari 2MRTU 960 and AOM 400). Randomization was stratified by disease type (schizophrenia or BP-I) and pharmacokinetic (PK) blood sampling schedule (robust or sparse, with the robust sampling schedule providing more frequent PK sampling timepoints [blood sampling schedules are presented in electronic supplementary Table 1]). A block size of 2 was used to generate the randomization sequence. As this was an open-label study, randomization sequence was not concealed internally. An Interactive Response System was used for participant randomization, and treatment assignments were based on a computer-generated code provided by Otsuka Pharmaceutical Development & Commercialization Inc. Access to the assignment codes was restricted.
All study treatments were administered by the investigators at the clinical trial sites. For participants stabilized on oral antipsychotic treatment, overlapping oral antipsychotic treatment was administered for 7 days after the first administration of Ari 2MRTU 960 or for 14 days after the first administration of AOM 400; there was no oral overlap for participants stabilized on AOM 400 (Fig. 1). Participants enrolled to a sparse sampling schedule could have been stabilized on oral aripiprazole, a non-aripiprazole oral antipsychotic, or AOM 400. For those stabilized on oral aripiprazole, they continued to receive oral aripiprazole for Days 1–7 (if randomized to Ari 2MRTU 960) or Days 1–14 (if randomized to AOM 400). For those stabilized on a non-aripiprazole oral antipsychotic, they continued to receive their current oral antipsychotic for Days 1–7 (if randomized to Ari 2MRTU 960) or Days 1–14 (if randomized to AOM 400). For those stabilized on AOM 400, no overlapping oral antipsychotic treatment was administered.
To be enrolled to the robust sampling schedule, participants must have been stabilized on a non-aripiprazole oral antipsychotic and must have demonstrated prior tolerability to aripiprazole. Participants enrolled to a robust sampling schedule discontinued their current oral antipsychotic and switched to 10–20 mg oral aripiprazole per day for the period of overlapping oral antipsychotic treatment with the first administration of study drug (overlap was 7 or 14 days depending on treatment group, with dose determined by the investigator).
In case of safety and tolerability issues, a one-time dose reduction was allowed for Ari 2MRTU 960 (to 660 mg) and AOM 400 (to 300 mg), with one-time subsequent increase back to 960 mg for Ari 2MRTU 960 or to 400 mg for AOM 400 allowed.
Participants enrolled to the robust sampling schedule and randomized to Ari 2MRTU 960 were housed in a trial site clinic for 21 days after the first and fourth doses of Ari 2MRTU 960. Participants enrolled to the robust sampling schedule and randomized to AOM 400 stayed in a trial site clinic for 21 days after the administration of the first, seventh, and eighth doses of AOM 400. Participants enrolled to the sparse sampling schedule were outpatients for the administration of all study drug doses but could be housed in the trial clinic for up to 21 days after the administration of the first study drug dose at the discretion of the investigator.
2.2 EndpointsPrimary safety and tolerability endpoints were evaluated throughout the 32 weeks of study duration and included reported adverse events (AEs); investigator’s assessment of the most recent injection site for symptoms of pain, swelling, redness, and induration; Visual Analog Scale (VAS) [29] scores for participant-reported rating of pain at the most recent injection site (range 0 [no pain] to 100 [extreme pain]); extrapyramidal symptoms (EPS), assessed by the Simpson–Angus Scale (SAS) [30], Abnormal Involuntary Movement Scale (AIMS) [31], and Barnes Akathisia Rating Scale (BARS) [32]; vital signs; electrocardiograms (ECGs); clinical laboratory monitoring (serum chemistry, hematology, and urinalysis); physical examinations; and suicidality (assessed by the C-SSRS) [33]. A schedule of safety and tolerability assessments is presented in electronic supplementary Table 2.
All AEs were coded by system organ class and the Medical Dictionary for Regulatory Activities (MedDRA) preferred term. A serious AE included any event that resulted in death; was life-threatening (i.e. the participant was, in the opinion of the investigator, at immediate risk of death from the event as it occurred); resulted in persistent or significant incapacity/disability or substantial disruption of the ability to conduct normal life functions; required hospitalization or prolonged hospitalization; resulted in congenital anomaly or birth defect; or resulted in any other medically significant events that, based on appropriate medical judgment, may jeopardize the participant and require medical or surgical intervention to prevent one of the previously listed outcomes. A treatment-emergent AE (TEAE) was defined as an AE that started after the first dose of study drug or an AE that continued from baseline and was serious, related to the study drug, or resulted in death, discontinuation, interruption, or reduction of the study drug dose.
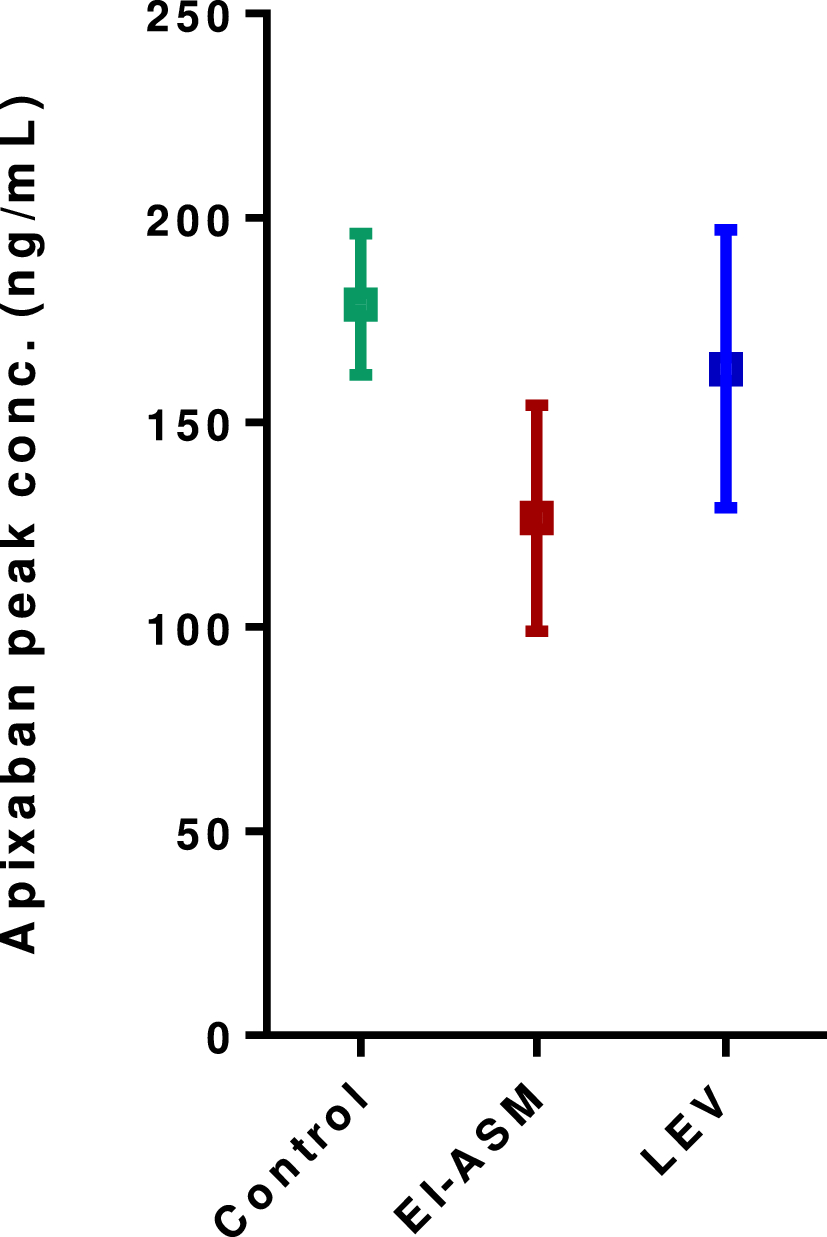
Primary PK endpoints were plasma concentration of aripiprazole 56 days after the fourth dose of Ari 2MRTU 960 (C56) and 28 days after the eighth dose of AOM 400 (C28), calculated based on PK data from participants enrolled to the sparse and robust sampling schedules; and area under the concentration–time curve (AUC) from Day 0 to 56 postdose (AUC0–56) after the fourth dose of Ari 2MRTU 960, or AUC from Day 0 to 28 (AUC0–28) after the seventh and eighth doses of AOM 400, calculated based on PK data from participants enrolled to the robust sampling schedule.
Secondary PK endpoints included maximum (peak) plasma concentration of aripiprazole (Cmax) and time to maximum (peak) plasma concentration (Tmax) after the fourth dose of Ari 2MRTU 960, and after the seventh and eighth doses of AOM 400 (in participants enrolled to the robust sampling schedule only) and AUC0–28 and AUC from Day 29 to 56 (AUC29–56) after the fourth dose of Ari 2MRTU 960 (in participants enrolled to the robust sampling schedule only). Peak-to-trough percentage fluctuation (PTF%) after the fourth dose of Ari 2MRTU 960 and after the eighth dose of AOM 400 were also evaluated (in participants enrolled to the robust sampling schedule only).
Secondary endpoints pertaining to efficacy were assessed by the Positive and Negative Syndrome Scale (PANSS) [34] and Clinical Global Impression—Severity (CGI-S) [31] for participants with schizophrenia, and by the Young Mania Rating Scale (YMRS) [35], Montgomery–Åsberg Depression Rating Scale (MADRS) [36] and Clinical Global Impression—Bipolar Version (CGI-BP) [37] for participants with BP-I. All participants were also evaluated using the Clinical Global Impression—Improvement (CGI-I) [31] and Subjective Well-being under Neuroleptic Treatment—Short Form (SWN-S) [38]. Efficacy outcomes were evaluated on Days 1, 29, 57, 113, 169, 197 and 225 or upon early termination of the study.
2.3 Statistical AnalysisStandard non-compartmental methods were applied to calculate the PK parameter values. The 90% confidence interval (CI) of the geometric means ratio (GMR) of C56 of aripiprazole after the fourth injection of Ari 2MRTU 960 (test) to C28 after the eighth injection of AOM 400 (reference) was calculated for all participants. Similarly, the 90% CI of the GMR of AUC0–56 of aripiprazole after the fourth injection of Ari 2MRTU 960 (test) to the sum of AUC0–28 after the seventh and eighth injections of AOM 400 (reference) was calculated for participants with robust PK sampling schedules. The GMRs and corresponding 90% CIs were derived from an analysis of variance (ANOVA) including treatment formulation and disease population as fixed effects for AUC and including treatment formulation, PK sampling and disease population as fixed effects for C56/C28. One-sided t-tests of treatment formulation within the specified ANOVA were performed to test the null hypothesis of the ratio of PK parameters for Ari 2MRTU 960 compared with the PK parameters for AOM 400 being ≤ 0.8.
It was estimated that a total of ≥ 100 participants (i.e., ≥ 50 per group) completing the trial would have ≥80% power to ensure that the lower limit of the 90% CI of the GMR of C56 after the fourth dose of Ari 2MRTU 960 (test) to C28 after the eighth dose of AOM 400 (reference) would be >0.80, assuming that the actual GMR of concentrations was 1.0. Among these 100 participants, at least 30 completers enrolled to the robust sampling schedule would provide ≥80% power to ensure that the lower limit of the 90% CI of the GMR of AUC0–56 of Ari 2MRTU 960 (test) to the sum of AUC0–28 values of AOM 400 (reference) after the seventh and eighth doses was > 0.80, assuming the actual GMR of concentrations was 1.15. Assuming a dropout rate of 34%, at least 152 enrolled participants were required to ensure at least 100 completers.
No data imputation was done for missing data in the PK analysis. The last observation carried forward (LOCF) method was used to impute missing data of efficacy assessment scales at post-baseline visits.
All randomized participants who received at least one study drug injection dose, regardless of any protocol violation, were included in the safety analysis. Time and dose of each study drug administration was recorded for each participant, along with information regarding any inappropriately administered dose. Treatment non-adherence criteria were defined as having a period of < 54 days between injections for Ari 2MRTU 960 and a period of < 26 days between injections for AOM 400.
The PK sample consisted of all dosed participants who had at least one evaluable aripiprazole PK parameter. For the primary PK endpoint analysis, only the completers with available values of the primary endpoints after the last scheduled injection were included.
All randomized participants who received at least one study drug dose and had at least one efficacy assessment were included in the efficacy analysis.
留言 (0)