
記住我
In this study, we evaluated a patient with a rare and severe neurological disease of suspected genetic etiology via whole genome sequencing and found two novel variants in trans configuration in the SPATA5 gene. Evidence for considering these two variants as causative is as follows. According to ACMG variant interpretation guidelines [35], we consider the frameshift variant fulfills the following pathogenicity criteria: (i) PVS1, since SPATA5 is predicted to have a LOF mechanism of disease by NMD according to in silico predictions in this study and others [2]; (ii) PM2, since no control population contains the variant according to several genomic studies, including ExAC [36], Gnomad [37] and Kaviar [38]; (iii) PM3, since the variant was detected in trans with the other missense (likely pathogenic) variant (sanger sequencing of parents confirmed the phase); and (iv) PP4, since the patient’s phenotype is highly specific for a disease with a single genetic etiology. Hence, PVS1, two PM and one PP are sufficient evidence for a “pathogenic” classification for this variant.
The missense variant fulfills at least, (i) PM2 (low population frequency), (ii) PM3, same rationale as before, (iii) PP2 (missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease), (iv) PP3, since PolyPhen [39], Sift [40, 41], Mutation Taster [42], FATHMM [43] and LRT [44] have deleterious predictions and phyloP scores (placental and vertebrate, meaning that only placentates or vertebrates were used to construct the alignments for the scores) [40] are greater than 1.6 (2.643, 8.738 respectively), hence it is a conserved region, suggesting pathogenicity, (v) PP4, same rationale as above. Hence, two PM and two or more PP are enough evidence for a “likely pathogenic” classification. However, we believe that we could add some evidence for PS3 (which would change the classification to “pathogenic”), since we have performed well-established in vitro functional studies supportive of a damaging effect on the gene product. In addition, there are some elements to believe PM1 could also apply, since the mutation is in the most functionally relevant domain of the protein (AAA2 domain).
From the ENCODE project, we obtained RNA-seq from different tissues, including skeletal muscle and spinal cord. Both of them could be relevant for the phenotype under study. Expression signal is observed all along the SPATA 5 gene, especially reads signal is observed toward the end of the transcripts supporting the expression of longer SPATA5 isoforms (Additional file 4: Fig. S4). Those transcripts are harboring both mutations.
We believe these variants are causal of the phenotype, based on clinical presentation being compatible with the phenotypes described for variants in this gene, various in silico analysis and mitochondrial function studies.
A recent report [5, 6] described several patients with heterozygous variants in the SPATA5 gene that were clinically suspected to present a mitochondrial disease, one of which presented defects in complex I and V in muscle. In this work, the authors also silenced SPATA5 expression in rat cortical neurons and observed altered mitochondrial dynamics, a decrease in mitochondrial length, as well as in the ATP/ADP ratio [5]. Another report [6] showed alterations in the levels of several mitochondrial proteins, as well as low enzymatic activity of respiratory complexes in a muscle biopsy of a patient carrying two pathogenic, compound heterozygous variants in the SPATA5 gene. Accordingly, we evaluated some parameters of mitochondrial physiology in blood cells from the patient and young control individuals. Our results showed an overall lower mitochondrial mass in monocytes obtained from the patient, compared to the control individual (Fig. 3). While the control is a young female, the patient is a male and some reports have shown that differences between sexes in mitochondrial mass could exist [34, 45, 46], though these appear to depend on the tissue and species. However, age-matched healthy pediatric controls are difficult to include in our studies, and a recent report shows a lack of significant differences in citrate synthase levels or mtDNA copies (classical markers of mitochondrial mass) in monocytes from men and women [47], supporting the comparison presented herein.
In platelets, oxygen consumption rate analysis revealed a significant increase in ATP-independent respiration, also known as “leak”, and decrease of the spare respiratory capacity in platelets from the patient, with respect to the healthy controls (Fig. 4). Respiratory indices were calculated and significantly lower coupling efficiency and respiratory control rate were found in platelets of the patient with respect to healthy controls (Fig. 4). The controls were two young females; however, previous reports show that sex and age do not affect platelet respiration [33, 48], supporting the validity of our results.
We also measured oxygen consumption rates in PBMCs. However, we did not find significant differences in mitochondrial respiratory parameters between the patient and female control individuals 18 and 20 years old, (Additional file 3: Fig. S3); only an increase in non-mitochondrial respiration. Since PBMCs respiration rates are reported to be higher in females than males [34], and to decrease with age [7], we compared the patient’s respiratory parameters with those of age-matched male controls from a recent study published by our group [7]. Several parameters indicative of mitochondrial function were significantly lower in the patient than in these controls, including basal respiration, maximum respiration and spare respiratory capacity. These results are similar to our previous observations in patients with mitochondrial disease, carrying mutations in mtDNA and nDNA [7].
We wondered if compensatory processes, such as an increase in mitochondrial biogenesis, could be taking place in PBMCs [7, 49,50,51,52]. To explore this possibility, we determined the number of copies of mtDNA in PBMCs, from the patient and control individuals, but found lower or similar levels in the patient. This result supports the decrease in mitochondrial mass, rather than the existence of a compensatory biogenesis program, in the patient. Nevertheless, other compensatory events such as an increase in catabolic pathways that provide substrates for the electron transport chain, cannot be discarded [53].
. In a recent report, we presented oxygen consumption rates for platelets and PBMCs from young control subjects and patients with mitochondrial disease, confirmed at the molecular level [7]. Interestingly, the values of most respiratory parameters (basal, ATP-dependent, maximum respiration and spare respiratory capacity) in platelets and PBMCs from our patient, carrying the variants in SPATA5, were similar or lower than the median values observed for the group of patients analyzed previously [7]. While the ATP-independent respiration in platelets (that denotes uncoupling), and the non-mitochondrial respiration (associated with reactive oxygen species) in both platelets and PBMCs from the SPATA5 patient were higher than those observed in our previous studies [7]. These results further support the value of studying mitochondrial function in blood cells for the diagnosis of mitochondrial disease. In sum, our observations suggest the SPATA5 mutation affects mitochondrial ATP synthesis, as well as the ability to respond to an increase in energy demand, in both platelets and PBMCs, probably due to a decrease in mitochondrial mass [17]. The results are in agreement with previous observations [5, 6] and constitute the first report of impaired mitochondrial mass and respiration in cells derived from a patient carrying mutations in the SPATA5 gene.
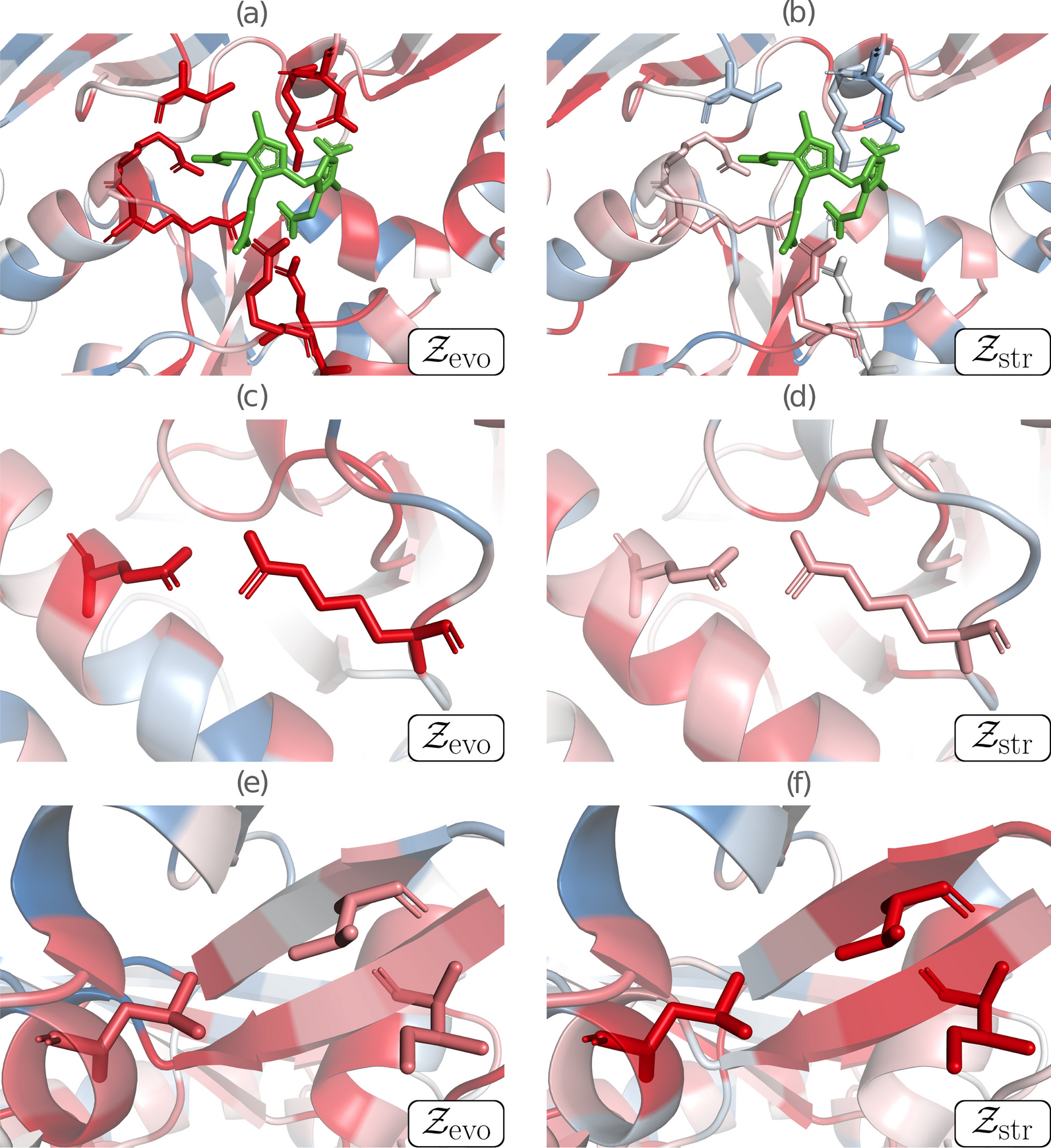
Structural characterization of AFG2, SPATA5’s homolog from baker’s yeast has shown it exists as a homohexamer [54]. Though some 100 residues shorter, AFG2 is reliably alignable to SPATA5 over its whole length. The reliable portion of a SPATA5 structural model obtained with AlphaFold [26], showed it to be superposable to the hexamer-forming domains of AFG2 (Fig. 6b–d). The poorly modeled N-terminal region (up to residue 350) reflects limited available sequences to train the deep-learning method (Fig. 5c). Indeed, this ~ 350 residue region looks confined to mammals, with very conserved sequence positions throughout the group. The six N-terminal regions of SPATA5 would be displaying a rich region for protein–protein interactions out of the hexamer. Three points support this hypothesis: (i) AF models with no assigned secondary structure often coincide with disordered or coil regions; (ii) a reliable portion of the N-terminal model is reminiscent of small beta barrel domains used as protein–protein interaction modules, e.g., as observed in Pleckstrin-homology domains [55]; (iii) finally, SPATA5 is thought to be a hub of protein interactions, engaging in many productive contacts [5].
Fig. 6
Protein structure modeling and comparative analyses of SPATA5 protein (ATPase family protein 2 homolog). A “Bottom” view of the hexameric experimental structure of SPATA5 homolog in S. cerevisiae (PDB 7KNU), colored by chain. B Rotated yeast hexamer superposed to model for SPATA5 homohexamer, manually built with AlphaFold monomers, in transparent gray cartoon. C Detail of the AlphaFold model for V766M SPATA5, color-coded from very high confidence (pLDDT > 90, dark blue) to very low confidence (pLDDT < 50, dark red). The C-terminal portion (Ile 677→end) affected by the frameshift mutation is shown as a transparent cartoon, coinciding with the very beginning of the second AAA + domain. The arrow depicts the connector affected by the V766M, represented as spheres. D Met766 substitutes a strictly conserved valine, affecting the beta 3 alpha2 connection composing the alpha/beta architecture of the AAA + domain (see “Discussion” Section)
We think that this finding can contribute to the use of whole genome sequencing as a diagnostic tool for challenging rare diseases, especially when no etiological orientation is given. Additionally, this kind of data might enhance the set of known mutations associated with different diseases. We think sharing this information is crucial for pushing genomic medicine further and improving diagnostic yields.
留言 (0)