1. IntroductionThe pathogenesis of type 1 diabetes (T1D) is fundamentally different from that of type 2 diabetes (T2D). While T2D in adults, is a metabolic disease resulting in insulin resistance, T1D generally occurring in adolescents, is an immune-mediated disease resulting in pancreatic β cell destruction, leading to insulin deficiency [
1,
2]. Although numerous therapeutic approaches have been tested experimentally [
3,
4], and several treatments are being used in the management of diabetic patients, no preventative strategies are currently available, partly due to the multifactorial nature of T1D [
5].T1D involves a strong genetic predisposition influenced mainly by human leukocyte antigen (HLA) class II genes, and to a lesser extent by HLA class I genes [
6], but the concordance rate of developing T1D among identical twins and siblings is as low as ~30% and 6%, respectively [
6,
7,
8]. This suggests that a combination of genetic and environmental factors such as infectious agents, drugs, dietary components, and gut microbiota may act in concert to trigger the disease in those affected [
9,
10,
11,
12]. Essentially, the prediabetic phase of T1D involves an autoimmune reaction causing the destruction of islet β cells accompanied by the appearance of antibodies to antigens such as insulin, glutamic acid decarboxylase, protein tyrosine phosphatase, Zinc Transporter 8 and cytoplasmic proteins in β cells [
13]. Exposure to microbial infections during this disease-developing stage in genetically predisposed individuals is believed to trigger clinically noticeable disease resulting in the destruction of residual islet β cells as infections are established [
5,
14].Of various microbial causes, a strong association exists between T1D and exposure to viruses, particularly enteroviruses such as Echoviruses, Coxsackievirus A and to a lesser extent, Rhinoviruses and Enterovirus 71 [
15,
16,
17]. However, epidemiological, clinical and experimental data points to group B Coxsackieviruses (CVBs) as major triggers [
6,
18]. Six CVB serotypes, CVB1 to CVB6 that infect various organs such as the heart, pancreas, liver, central nervous, and gastrointestinal systems have been identified [
19,
20,
21]. While all serotypes can cause pancreatitis, CVB3 is generally implicated in myocarditis [
22,
23,
24], and CVB1 and CVB4 are known to be associated with insulitis, thus acting as cofactors for the development of T1D [
25,
26,
27]. The reason for such differential disease phenotypes remains obscure since CVBs require Coxsackievirus-Adenovirus Receptor (CAR) to enter the target cells [
28]. Importantly, endocrine cells of the pancreatic islets highly express CAR, providing an explanation for the tropism of CVBs [
29,
30]. Of note, identities between all six CVB serotypes are 76–80% and 86–91% at the nucleotide and amino acid levels, respectively. Thus, coordinated expression of various viral and host factors may determine damage to a specific tissue or cell type. It is also possible that the expression of specific isoforms of CAR may be necessary for different CVB serotypes to infect various cell types [
31]. Nonetheless, the strong association between the occurrence of T1D and two CVB serotypes (CVB1 and CVB4) presents an opportunity to develop vaccines that may significantly impact the occurrence of T1D.To that end, we made efforts to develop vaccines for CVBs and identified one live-attenuated CVB3 vaccine strain, designated mutant (Mt)10, which can prevent infections caused by homologous (CVB3) and heterologous (CVB4) strains [
32,
33]. As described elsewhere, the Mt10 virus vaccine prevented both myocarditis and pancreatitis by inducing neutralizing antibodies (nAbs) and antigen-specific T cell responses in challenge studies [
32,
33]. By extending these observations, we report here that the Mt10 vaccine virus could prevent CVB4-accelerated T1D in the non-obese diabetic (NOD) mice by inducing cross-reactive nAbs. As expected, however, the spontaneous progression of T1D in vaccinated mice was not altered. 3. Results and DiscussionWe recently reported the creation of a novel live-attenuated CVB3 vaccine candidate, Mt10, that offers protection against both homologous (CVB3) and heterologous (CVB4) serotypes of CVB [
32,
33]. CVBs affect various organ systems [
19,
20,
21], and although similar diseases are induced by multiple serotypes, their disease severities may vary in susceptible mouse strains such as A/J, BALB/c, or NOD mice [
41,
42,
43]. For example, while CVB3 could induce severe myocarditis in A/J and BALB/c mice [
44,
45], pancreatitis could be induced to a comparable severity by CVB1, CVB3, and CVB4 [
46,
47,
48]. Conversely, CVB4, despite infecting the heart tissue, has little myocarditis-inducing ability [
32,
48]. However, both CVB1, CVB4 and to a lesser extent CVB3 have been shown to trigger T1D in the NOD mouse model [
41]. These variations appear to be due to the tissue tropism of different CVB serotypes [
29,
30]. For example, CVB4 infects pancreatic β cells, whereas the exocrine pancreatic acinar cells are infected by CVB3 [
49]. Nonetheless, because of a high degree of similarity among all three serotypes (
Table S1), there may be an induction of cross-reactive immune responses. Consistent with this prediction, our vaccine candidate, Mt10 vaccine virus bearing the CVB3 backbone, was found to induce robust cross-protective immune responses against CVB4, and to a lesser degree, CVB1 in A/J mice. However, the vaccine’s ability to influence T1D development in NOD mice was not investigated. Thus, we sought to test the hypothesis that the Mt10 vaccine could prevent accelerated T1D development triggered by diabetogenic CVB serotypes.To address our hypothesis, we chose CVB4 since the Mt10 vaccine virus could induce significant levels of cross-reactive nAbs to CVB4 in A/J mice [
32]; and CVB4 was readily available for challenge studies. Of note, NOD mice begin to show inflammatory infiltrates with loss of pancreatic β cells around 8 weeks of age [
50]. At about 12 to 15 weeks, these mice spontaneously develop insulitis and hyperglycemia [
51,
52], which can be aggravated to a greater severity upon exposure to CVB4 [
27]. Furthermore, reports suggest that CVB infections can delay the diabetes process in young NOD mice, likely due to a non-specific viral effect on the murine immune system [
53], whereas the same infections can accelerate the disease in older mice [
41]. We used 6-week-old mice and immunized them with Mt10 twice, at 2-week intervals; 7 days later (equivalent to 9 weeks), animals were challenged with CVB4. At termination, animals had reached 13–14 weeks of age (
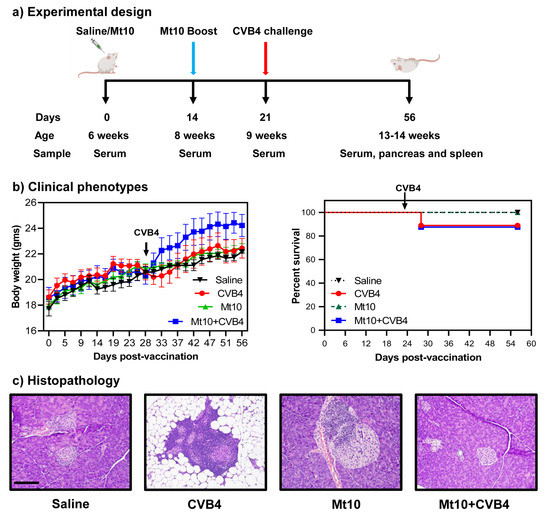
Figure 1a). All animals gained weight over time, which is typical of healthy mice. Clinically, infected mice lost weight ~1-week post-infection and thereafter, but the weight loss was not substantial (
Figure 1b, left panel). Likewise, none of the animals in any of the groups succumbed to disease. However, one mouse each in the CVB4 and Mt10 + CVB4 groups (~12%) died from virus infection (
Figure 1b, right panel). Otherwise, animals in all groups appeared clinically normal during the 56-day observation period.To further evaluate the disease-protective ability of the vaccine virus, we analyzed pancreata collected at termination for insulitis and other changes by H. and E. staining [
33,
54] (
Figure 1c,
Table 1). First, we noted mild insulitis in saline recipients (40%), which was an expected finding since NOD mice develop T1D spontaneously around 12–15 weeks of age [
51,
52]. Second, 89% of CVB4-infected mice had severe insulitis marked by the destruction of islets and the replacement by adipose tissue. Such an aggravation is consistent with previous reports that CVB4 infection could potentiate the progression of T1D [
55]. Third, although insulitis was noted in 57% of vaccinated animals, vaccinated animals challenged with CVB4 had a significantly lower incidence of insulitis (13%) (
Table 1). While a similar trend was noted with peri-ductule inflammation, the occurrence of peri-insulitis was not different across the groups, raising the question of whether pancreatic pathology could be correlated with the development of T1D and virus multiplication.In this direction, we measured blood glucose levels once a week. We noted that none of the saline- or vaccine-recipients developed T1D (
Figure 2a). In contrast, 70% of mice in the CVB4-alone group became diabetic, whereas only one mouse (12%) from Mt10 + CVB4 developed diabetes (
Figure 2a). Additionally, we tested whether insulin antibodies followed a similar pattern. We chose to measure antibodies to insulin in our studies since they are commonly detected in T1D patients [
56,
57]. Nonetheless, standardizing insulin antibody assays in mice has been consistently debated [
58,
59,
60,
61], and this limitation was previously circumvented by developing a sensitive competitive europium insulin autoantibody assay [
37]. Essentially, in this assay, because europium chelate has a longer fluorescence lifetime than traditional fluorophores, introducing competition for insulin greatly reduces the background signals that otherwise have been a technical hurdle with traditional insulin ELISAs [
37]. The analysis revealed that only the CVB4-infected mice (at day 56) and 1 out of 5 vaccinated mice had elevated levels of insulin autoantibodies (
Figure S1). In contrast, control mice and vaccinated mice challenged with CVB4 (at day 56) had relatively low levels of insulin antibodies, but no significant differences were noted between groups. Of note, it is common to evaluate at least two autoantibodies to determine the progression of T1D in clinical settings [
7,
62], which we have not done in our studies.We next investigated if the vaccine-mediated protective effects are attributable to prevention of CVB4 infection by examining pancreata for viral RNA. The RT-qPCR analysis indicated that vaccinated mice as well as those vaccinated/challenged with CVB4 had negligible or no viral nucleic acids in their pancreata (
Figure 2b). A relatively increased expression of viral RNA was detected in mice infected with CVB4 (
Figure 2b), but the differences were not significant. This data suggest that virus persistence may not be necessary to accelerate the disease process and that by preventing infection, the vaccine virus might have suppressed the development of T1D in challenged animals.To mechanistically evaluate whether vaccine-mediated T1D protection might be due to induction of nAbs, we performed a live virus neutralization assay on sera collected before and after vaccination (at days 14, 21 and 56;
Figure 3). Expectedly, none of the animals in the control group had any detectable nAbs for any of the CVB serotypes tested (
Figure 3, top left panel). Similar analysis revealed that mice vaccinated with Mt10 had nAbs against CVB1, CVB3, and CBV4 in the decreasing order of CVB3/CVB4, and CVB1 (
Figure 3, top right panel). Of note, nAb levels for CVB1 were similar across all time points in vaccinated mice with GMTs ranging from 289 to 343, whereas nAb levels for CVB3 reached a GMT of 3044 post-prime, and peaked on day 21 (post-boost) with a GMT of 22,334, and reached closer to the pre-boost steady levels (GMT 6891) by day 56 (
Figure 3, top right panel). Consistent with our previous observations [
32], the high nAb titers against CVB4 associated with Mt10 vaccine peaked at the post-boost time point (day 21) with a GMT of 8611 and were stably maintained thereafter (GMT 2827) (
Figure 3, top right panel). We next compared the nAb titers between vaccinated and vaccinated/CVB4-challenged groups at day 56, leading us to note that the titers of nAbs for CVB1 remained similar in both groups with a GMT of 290 for vaccinated and GMT of 253 in CVB4-challenged group (
Figure 3, bottom panel). Further analysis revealed a significant increase in nAbs for CVB3 (GMT 36,491) in the CVB4-challenged group (
Figure 3, bottom panel), indicating that CVB4 could increase the cross-reactive vaccine response to CVB3. Although such a trend existed for CVB4 (GMT 9123) in the challenged group, the differences in nAb titers were not significant between vaccinated and challenged groups (
Figure 3, bottom panel). Yet, it was critical to determine the nature of antibody isotypes produced in each group.We evaluated the antigen specificity of antibodies generated against CVB4 by using the VP1 of CVB3 and KLH as positive and control antigens, respectively, as described previously [
32,
33], since a high degree of similarity (~90%) exists between VP1s of CVB3 and CVB4. We made two major observations using the sera collected on days 0, 14, 21, and 56 (
Figure 4). (i) Total Ig levels were significantly elevated in both the Mt10 (p ≤ 0.001) and Mt10 + CVB4 groups (p ≤ 0.01) as compared to the saline group (
Figure 4). The Ig levels persisted all through the 56-day experimental period. Likewise, animals in the CVB4-alone group revealed significantly elevated levels of Ig (p ≤ 0.05). These data suggest that the NOD mice could respond to multiple CVB serotypes and generate immune responses. (ii) Patterns similar to those described above were noted for IgG2c and, to a lesser extent, IgG2a (
Figure 4). Otherwise, no alterations were noted in other isotypes (IgM, IgG1, IgG3, IgA, and IgE). Thus, we concluded that vaccine-induced protection might have occurred through the generation of predominantly IgG2c nAbs cross-reacting with CVB4. This protective ability of the vaccine may involve the mediation of virus-specific T cell responses since T cell cytokines are critical for isotype switching. For example, IFN-γ promotes IgG2c [
63,
64]. In that direction, we tested for various anti-viral (
Figure S2) and T helper subsets (
Figure S3) cytokines/chemokines in the serum samples collected on days 0 and 56. However, none of the cytokines tested showed any significant differences between groups. It is possible that cytokine analysis in the blood compartment at the late time points is unlikely to yield informative data. We drew similar conclusions by evaluating various B cell subsets in the splenocytes, such as B cells (CD19+B220+), class-switched B cells (IgM−IgD−), and germinal center cells (CD95+CL7+) (
Figure S4), including the possibility that Mt10 vaccine could induce the regulatory T (Treg) cells (CD4+FoxP3+) because CVBs have been shown to induce Treg cells [
65,
66,
67]. The analysis revealed no major differences between groups, except that the Ig class-switched cells tended to occur more in the CVB4-alone group, followed by the vaccine and vaccine/CVB4-challenged groups. Likewise, an increasing trend was noted in the vaccine group for the frequencies of Treg cells when compared to the saline group [p = 0.06] (
Figure S4). Taken together, our data suggest that cross-reactive nAbs may have led to the prevention of CVB4 infection, thus contributing to vaccine-induced protection against CVB4-accelerated T1D. This notion is supported by the finding that the CVB4-alone group had exacerbated T1D (
Figure 2).Given the importance of T1D, the use of vaccines to prevent the disease has been discussed in the literature [
68,
69,
70,
71], but questions may arise as to which viruses can be targeted and how, what the target population for the vaccine is if vaccines become available, and what the impact might be. There are no easy answers to these questions, primarily because T1D has been associated with a variety of viruses [
18,
72]. Based on serological evidence, however, the major candidates appear to be CVBs at a global level [
73,
74]. Furthermore, although it is hard to prove the cause-and-effect relationship in humans, CVBs could cause diabetes in monkeys [
75], and studies from the pre-clinical NOD model suggest that CVBs could trigger T1D in genetically susceptible individuals [
52]. In support of this proposition, enteroviruses and their signatures (antibodies or nucleic acids) have been detected in T1D patients, including genetically at-risk infants [
76,
77,
78]. Thus, a suggestion has been made that vaccines may prevent the initiation of infections when given to genetically susceptible individuals at an early age prior to the start of the β cell damaging process, which would limit T1D development in vaccine recipients [
79,
80]. However, accumulated literature suggests that T1D can be seen in patients without previous T1D history, implying that vaccinating broader populations may be appropriate [
81,
82]. Furthermore, infection of β cells with viruses can occur at any age, and T2D patients could have T1D as enteroviruses have been detected in T2D patients at a higher rate than in unaffected controls [
15,
83,
84]. It has also been proposed that vaccine viruses may induce disease-protective Treg cells, as shown in NOD mice exposed to CVB infections [
65,
66]. Therefore, the use of anti-viral vaccines has been estimated to prevent at least 50% of T1D cases [
18], but questions arise as to their desired characteristics.It is not practical to generate serotype-specific vaccines to prevent T1D since multiple CVB serotypes can trigger the disease [
6,
18]. For having successfully developed effective vaccines against polioviruses [
15,
85] and enterovirus 71 [
70,
86], generation of similar vaccines for T1D in relation to CVBs is possible. In that direction, formalin-inactivated non-adjuvanted monovalent CVB1 vaccine was shown to prevent CVB1 infection and also CVB1-induced T1D development in NOD mice and the transgenic mice expressing suppressor of cytokine signalling-1 on the NOD background [
87]. Likewise, formalin-inactivated whole CVB4-E2 was shown to delay the onset of T1D in NOD mice, but autoantibodies to several beta-cell antigens were elevated potentially resulting from cross-reactivity [
88]. Recently, a hexavalent inactivated vaccine involving all six serotypes of CVB has been tested in mice and nonhuman primates [
89]. Although this vaccine was effective in preventing CVB infections by producing nAbs, antigen specificity and characterization of antibody isotypes were not investigated. Our vaccine is a live attenuated virus able to induce both antibody and T cell responses. While we have demonstrated the former, we could not analyze antigen-specific T cell responses in NOD mice due to a lack of relevant tools. However, the possibility remains that virus-reactive T cells can be generated in vaccine recipients because antibody production—as expected in NOD mice—was almost entirely skewed toward the IgG2c response that requires IFN-γ from T cells [
63,
64]. Conversely, CVBs are RNA viruses and therefore prone to mutations [
90,
91], so it is possible that the live vaccine viruses may revert to virulence. To overcome this possibility, we are generating virus-like particles (VLPs) from Mt10 with the expectation that their structural proteins could induce protective responses similar to live Mt10 virus. In support of this notion, the VLP-based vaccine for CVB3 was shown to induce high nAbs in mice, but its ability to prevent T1D development was not investigated [
92].In summary, we have demonstrated that the monovalent Mt10-CVB3 vaccine virus could protect against CVB4-accelerated T1D in NOD mice. Since multiple CVB serotypes could induce similar organ-specific diseases, as is the case with CVB1 and CVB4 in the development of T1D [
25,
26,
27], the Mt10 vaccine has the potential to produce cross-reactive nAbs for other serotypes. We had previously demonstrated that the Mt10 vaccine could prevent myocarditis induced by CVB3, as well as pancreatitis induced by both CVB3 and CVB4 in A/J mice [
32,
33]. Thus, we expect that the Mt10 vaccine could be used to prevent infections caused by multiple CVB serotypes that induce different organ-specific diseases. While such investigational studies in the mouse models provide a proof of concept, for a greater validation, vaccine efficacies are to be tested in the non-human primate models of T1D [
93,
94].

留言 (0)