In different models of breast cancer and melanoma, it has been observed that GK-1 has an antitumor effect decreasing angiogenesis, metastasis, as well as tumor growth, which has been related to the modulation of different factors such as the decrease of different cytokines associated with malignancy such as basic fibroblast growth factor (bFGF), granulocyte macrophage-colony stimulating factor (GM-CSF), tumor necrosis factor alpha (TNF-α) or chemokine (C-X-C motif) ligand 9 (CXCL9), increasing interleukin (IL)-6 and IL-12 levels [
5]. It has also been found that GK-1 induces anti-tumoral cluster of differentiation 8 (CD8) T cell response associated with the downregulation of the programmed cell death protein 1 (PD-1)/ programmed death-ligand 1 (PD-L1) pathway [
10]. The increase of ROS and OS are conditions associated with mitochondrial damage and dysfunction that have been linked to cancer development and its elimination [
36]. Considering this, we wondered if GK-1 targets the redox state, mitochondrial bioenergetics, and autophagy process. We found that in the TNBC mice model, GK-1 induces OS and oxidative damage since an increased mitochondrial H2O2 production, GSSG, and protein carbonyl contents, and a reduction of the antioxidant activity of catalase, GSH content, and the GSH/GSSG ratio (
Figure 2,
Figure 3 and
Figure 4) was found. This prooxidant effect of GK-1 is similar to that caused by peptides from the venom of the marine cone snail Conus vexillum, which induces OS by increasing ROS and reactive nitrogen intermediates as well as decreasing the activities of antioxidant enzymes catalase and SOD, causing oxidative damage such as lipid and protein oxidation in carcinoma cells. This induction of OS and its cytotoxic effects confer to venom-anticancer properties [
37]. GSH is the main antioxidant in the mitochondria and helps to maintain mitochondrial homeostasis, so when a decrease in GSH content and H2O2 production increase occurs, OS is promoted, which induces mitochondrial oxidative damage and a decrease in cell viability [
38]. Thus, the OS induced by GK-1 would compromise tumor growth and metastasis as a consequence of the oxidative damage that occurs in the mitochondria. We found that tumors treated with GK-1 peptide decreased their respiratory S3, S4o, P, and RC parameters (
Figure 5B–E). The S3 respiration corresponds to the oxygen consumption in the presence of all the substrates that allow the mitochondria to perform OXPHOS, and we found that this decreases significantly with GK-1. The S4o refers to cellular oxygen consumption that does not correspond to OXPHOS, which is also decreased by GK-1 treatment. Moreover, P, linked to ATP production and RC, also decreases, showing that mitochondria cannot use adequate oxygen to produce ATP. It should be noted that if the RC index is low, it indicates that mitochondria proton leak processes increased, and the oxygen is being used for other processes not associated with the production of ATP. For instance, one of these oxygen processes could be used by ROS production [
39], which agrees with the higher levels of mitochondrial H2O2 production. These results suggest that GK-1 induces mitochondrial dysfunction, where mitochondria do not use oxygen efficiently for ATP production. Moreover, we observed that tumors treated with GK-1 have fewer ATP synthase subunit levels, which compromises tumor cell respiration, demonstrated by the significant decrease in S3, P, and RC (
Figure 5B–D). The cell growth is dependent on mitochondria since this organelle provides not only energy in ATP form but metabolites that induce cell growth [
40]. Regarding the latter, we found that ATP synthase and VDAC1 levels are decreased in GK-1-treated tumor cells (
Figure 7), which agrees with the reduction of tumor, as ATP synthase and VDAC1 are essential proteins in mitochondrial metabolism and ROS-mediated cell death [
34,
41]. On the other hand, the results of ΔΨm analysis show that GK-1 decreases it in S2, S3, and S4o, indicating that the mitochondria are not capable to maintain the polarization status. In the case of S3, the ΔΨm loss explains the decreased ATP production capacity in mouse TNBC. The loss of ΔΨm is closely associated with cell viability since a decrease in the potential can induce cell death [
42]. These effects agree with the effect of CIGB-552, a synthetic anti-tumor peptide developed by The Center for Genetic Engineering and Biotechnology (CIGB), which decreases SOD1 activity, reduces cellular antioxidant capacity, and promotes ROS production. The latter induces more damage to mitochondria and increases the oxidative damage of lipids and proteins in tumor cells, leading to apoptosis-mediated cell death. In this study, authors show that CIGB-552 effect reduces tumor growth of human colon tumor xenografts [
43]. One of the processes that allow the elimination of proteins and organelles damaged by OS is autophagy, so by measuring the markers of this process, we can assume that tumor cells could be protecting themselves from OS damage. However, we found that GK-1 increases p62 autophagy marker, indicating that GK-1 stops autophagic flux (
Figure 9). Yan et al. showed that invading human primary non-small cell lung cancer (NSCLC) exhibits higher autophagic flux than cancer cells inside the tumor body; when this autophagy flux is stopped, tumor invading capacity decreases [
44]. It is important to note that we only measured p62 as a marker of autophagic flux. Measuring LC3 turnover assay together with electron microscopy techniques will define whether GK-1 reduces autophagic flux or not. Moreover, the measure of additional mitochondrial mass markers such as mitochondrial DNA or imaging mitochondrial network by electron microscopy would help to overcome these limitations of our work. Even with these limitations, our results make an approximation of the effect of GK-1 on autophagic flux and mitochondrial mass, which suggest that the arrest of autophagic flow by GK-1 could decrease the invasion and metastasis capacity of 4T1 cells, as has been shown by Torres-García et al. [
5]. However, more investigation is necessary to conclude these effects. Thus, we found that GK-1 induces OS, mitochondrial dysfunction characterized by ΔΨm loss, uncoupling, and higher ROS production, as well as a probable disruption of autophagic flow, which could compromise tumor cell viability. It is also probable that GK-1 directly affects mitochondrial homeostasis, inducing mitochondrial dysfunction, which in turn would promote ROS production and OS, having positive feedback in ROS generation, which is reinforced by the decreased catalase activity during GK-1 treatment. The latter, together with the stop of autophagy flux induced by GK-1 will reduce tumor growth and metastasis (
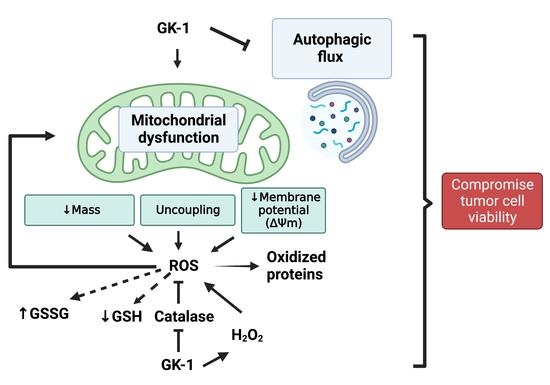
Figure 10) [
5]. Notably, it is important to highlight that in preclinical studies, GK-1 peptide was safe as the absence of subchronic toxicity and mutagenicity was found [
45], accounting for these effects only in tumoral tissues. Discovering the exact mechanism of how GK-1 induces mitochondrial dysfunction deserves further investigation because this could open up new avenues for applying drugs that exert tumor elimination.



留言 (0)