
記住我


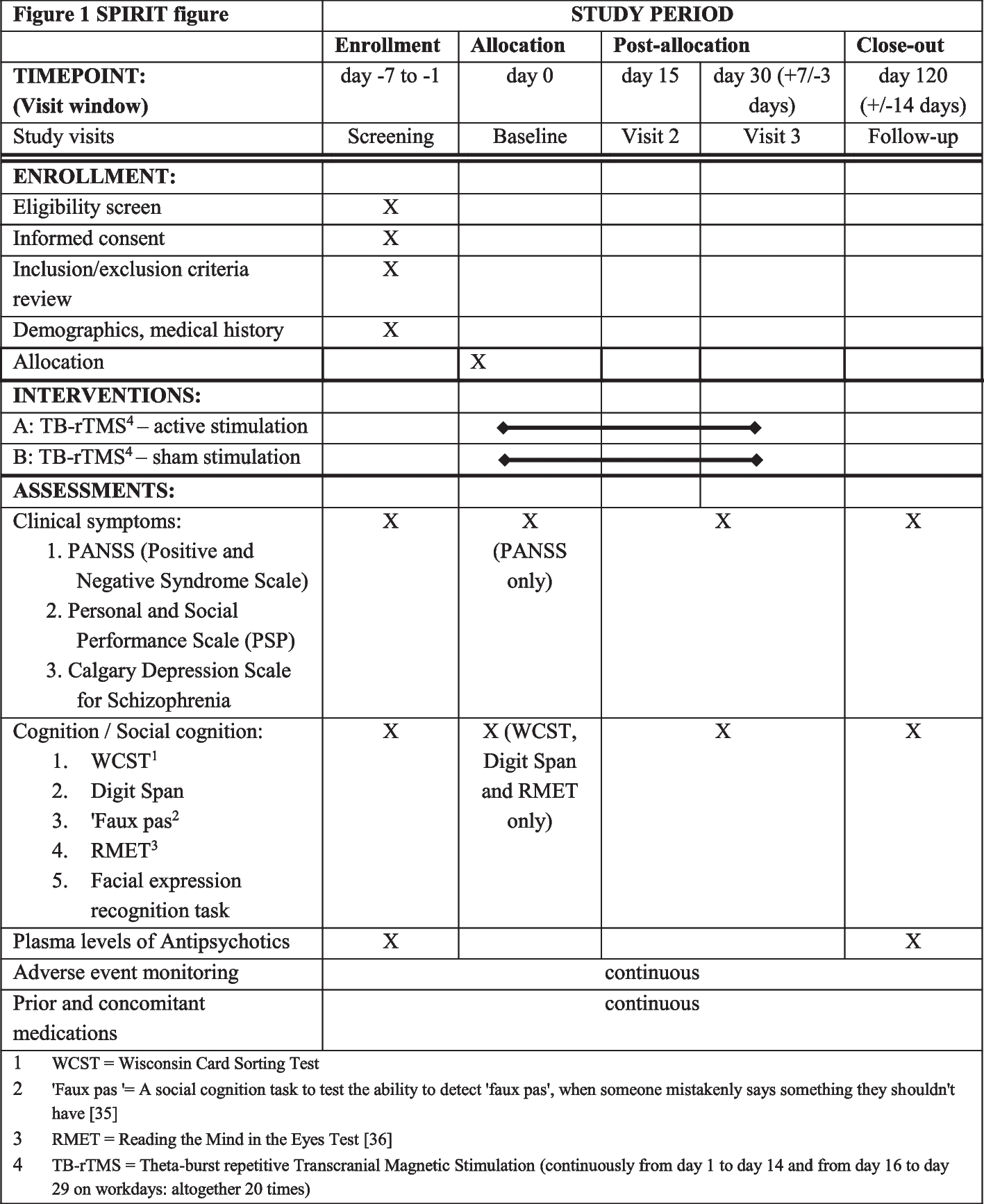

The trial protocol is identified as version n°2.0 of 11 December 2019. It adheres to Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) criteria; the SPIRIT Checklist can be found as Additional File 1: Table S1.
DesignThis noninferiority, prospective, randomised, controlled, single-blind, two-group study will be conducted in the emergency subunits of the Nantes University Hospital. Eligible patients with irreversible pulpitis in the first or second mandibular molars requiring emergency dental care will be included in the study. Diagnosis will be based on clinical and radiographic examination. Two parallel groups will be randomised at a ratio of 1:1 so that patients will either receive (1) intraosseous anaesthesia or (2) inferior alveolar nerve block anaesthesia. This manuscript used the SPIRIT reporting guidelines [30].
Involvement in the design of the protocolNo members of the public or patient groups were involved in the design of the protocol.
ParticipantsAdult patients seeking emergency consultation at the dental department of Nantes University Hospital will be included in the study according to the following criteria:
Presence of spontaneous and/or diffuse pain with severe sensitivity to cold and/or heat tests in the first or second mandibular molar
Patients over 18 years old (of any gender)
Patients in good health, with no medical history (ASA 1 score) and no drug treatment for cardiac rhythm disorders (antiarrhythmics, beta-blockers)
Patients able to give his/her oral consent.
Affiliated with a health insurance
Agree to be contacted by phone 72h after the emergency treatment
Participants will not be included if they present at least one of the following non-inclusion criteria:
Contraindications to anaesthesia with vasoconstrictors
Diagnosis of irreversible pulpitis of third mandibular molars, reversible pulpitis, acute apical periodontitis, periodontal lesion of endodontic origin or dentin syndrome
Non retainable tooth requiring extraction
Vital tooth serving as an abutment in fixed prosthesis
Patients under the age of 18
Adult patients who do not understand French or have refused to consent will not be able to participate
Pregnant or breastfeeding women
OutcomesThe main outcome of this study is to compare the time required in minutes for the cardiovascular parameters to return to their initial values between the two techniques: ICA with the QuickSleeper™ system versus the reference technique: inferior alveolar nerve block (IANB). The timing of the evolution of the parameters is based on 3 phases: “baseline” from T0 to T+4 minutes, “anaesthesia” from T+4 to T+8 minutes, and “pulpotomy” from T+8 to T+14 minutes. The return to baseline values will be defined as a success if 3 successive values of heart rate and blood pressure (systolic and diastolic) are identical to those recorded before injection. On the other hand, if there is an absence of 3 successive values of these two parameters, it is defined as a failure.
Secondary outcomes include:
The O2 measurement performed during the 3 phases
The efficacy of the anaesthesia with a VAS (visual analogue scale) of 0 and that the pulpotomy was completed successfully: the patient is asked about his or her perception of pain before the operation at D0 and D+1, D+2, and D+3
Post-operative care
Investigation of factors associated with delay in recovery of cardiovascular parameters (heart rate, dental anxiety, age, gender, and use of analgesics in the 6 h before surgery)
Study timelinesAll patients seeking emergency consultation at the dental subunits of the Nantes University Hospital will be screened for the selection criteria. If a patient meets all criteria, he or she will be informed by an investigator and the investigation team of study technicians. He or she will be handed an information letter that comprises the objectives, methods, follow-up risks and restrictions of the trial. The patient’s oral consent is required before the start of any intervention and will be under the responsibility of the investigator to obtain consent. Thus, the patient’s consent will be obtained by the investigators only (Fig. 1).
Fig. 1
Flowchart of a trial evaluating the noninferiority of ICA QuickSleeper™ system versus IANB anaesthesia for the pain management in the emergency care of irreversible pulpitis in mandibular molars at the Nantes University Hospital
The investigator in charge of the emergency room will proceed to randomisation (see below) using a secure computer program called Ennov Clinical and will conduct the emergency endodontic treatment. The patient will then be contacted by a research assistant 72h following the intervention by telephone to assess the pain perception and if any secondary effects of the anaesthesia occurred, as depicted in Table 1. Part or all of the study may be discontinued permanently or temporarily by the decision of the ANSM, the CPP, and the Study Sponsor (Nantes University Hospital).
Table 1 Schedule of enrolment, interventions, and assessments during COQ study protocolAuditingThe audit process will be done independently from investigators and the sponsor.
InterventionsThe patient will be blinded to the affected group and will fill out the first questionnaire which is the French version of Corah’s dental anxiety scale [31] to assess his or her anxiety about dental care. Then, the patient will be placed with a cuff on the arm, and a pulse oximeter on the other finger connected to a blood pressure monitor to monitor the cardiovascular parameters (heart rate, systolic and diastolic blood pressure, and oxygen saturation) throughout the intervention.
Sequence generationEach patient, regardless of the study group allocated, will receive 1.8 mL of 4% articaine 1:100 000 for 2 min. The evaluated intervention consists of the administration of intraosseous anaesthesia using the QuickSleeper™ (DHT, Cholet, France) system, according to the manufacturer’s recommendations, in the “ICA” group. A plastic syringe with a cartridge of 4% articaine 1:100,000 epinephrine will be used in the “IANB” group. The emergency treatment will be a pulpotomy. The tooth will be isolated with a rubber dam and all carious tissue and pulpal parenchyma will be removed. Pulpal bleeding will be controlled using a 2.5% sodium hypochlorite and the site will be covered with calcium hydroxide and a temporary filling. At the end of the intervention, all patients, regardless of the randomisation group, will be given two types of antalgics (paracetamol 1 g or paracetamol 600mg/codeine 50 mg) according to preoperative pain declaration (VAS) and recommended to take them only if they experience pain, according to the good clinical recommendations.
RandomisationPatients will be randomly assigned to one of the two arms: “ICA” and “IANB” at a ratio of 1:1. The randomisation list will be computer-generated by the study statistician and the randomisation process will be centralised through a secured clinical data processing website managed by Ennov© Clinical system software (Ennov, Paris, France). The investigators will enter the patient’s information (initials, age, and date of birth mentioning the month and year only), and the software will provide the patient’s unique allocation number and randomisation group.
Allocation concealment mechanismAs it is a randomised controlled study, the patients are blinded. The unblinding occurs if a patient decides to leave the protocol prematurely. The best care will be provided by the investigator and the pulpotomy will be performed under the best conditions to relieve the patient’s pain.
Determination of sample sizeThe sample size calculation was based on the noninferiority hypothesis in a pilot study that there will be an expected mean difference of 1 min 30 s between the two anaesthetic techniques with regard to the time taken for the values to return to the initial ones. According to the reported results from the few studies available, the proportion of patients having successful anaesthesia and better pain control in the reference group (intraosseous anaesthesia) would be 93%. The noninferiority margin was defined as 20%. The significance was set at 1.7% for the 3 main criteria (heart rate, systolic and diastolic blood pressure) and 5% for the secondary criteria. Thus, using a bilateral test with an epiR package 0.98-87 (Biostatgv), 72 patients (36 patients per arm) will be included in the study.
Statistical analysisThe analysis will be based on intention-to-treat (ITT) using the “missing=failure” strategy. A robustness analysis, supported by the per-protocol approach, will be carried out if the patients present with protocol deviations (error of randomisation). Qualitative variables will be described by the percentages and numbers of each modality. Quantitative variables will be described by means and standard deviations. Confidence intervals at 95% will be presented.
A paper case report form (CRF) will be created for each patient inclusion for data collection and will be based on the establishment of a clinical database. All information required by the protocol must be provided in the CRF, necessary for statistical analyses.
To assess the primary endpoint (time to return to baseline heart rate and blood pressure), the mean values will be estimated by the Kaplan-Meier method (survival curves) and compared using Log-Rank tests. To assess the secondary endpoint (time to baseline O2 and change in measurements over time), the 2 arms will be compared in the same way as the heart rate and blood pressure parameters. Univariate and multivariate Cox models will be used to investigate the factors associated with the time to the baseline of cardiopulmonary constants. The difference in pain between D0 and D+3 will be compared between the 2 groups a Student’s t-test or a Wilcoxon test. Data for the analysis are collected from the first questionnaire (French version of Corah’s dental anxiety scale), the three phases of cardiopulmonary monitoring, the VAS assessment, and the postoperative questionnaire given to the patient at the end of the procedure. The information recorded on the postoperative questionnaire will be collected by a telephone call 3 days after the procedure. All the data collected will be typed and inserted into an Excel spreadsheet for data entry in a secure computer network of Nantes University Hospital. The investigative team, mainly the investigators as well as the study technicians involved in this study protocol will have access to the data collected.
In regard to the post-trial care, the patients will either continue their dental care with their appointed dentist, or in the case where they do not have one, they will be following appointments at the Nantes University Hospital.
A dissemination policy concerning the statistical data will be implemented to ensure that the official statistics are consistent, reliable, and comparable.
Protocol violationsAll protocol violations occurring after randomisation will be listed in the clinical report form (CRF) and tabulated by the subject. The final assignment of participants to the per-protocol analysis will be decided at a blinded protocol review meeting before locking the database.
Adverse eventsConsidering the low risks and constraints of this study, no adverse event related to the trial is expected. In addition to the measurements of blood pressure and heart rate variations, which are the objectives of the present protocol, the suspected AEs related to the administration technique or anaesthesia will be traced in the CRF and analysed according to the study schedule by internal monitoring. They will not be notified to the sponsor, even if they are serious. Any adverse event is linked to a failure of anaesthesia.
留言 (0)