
記住我


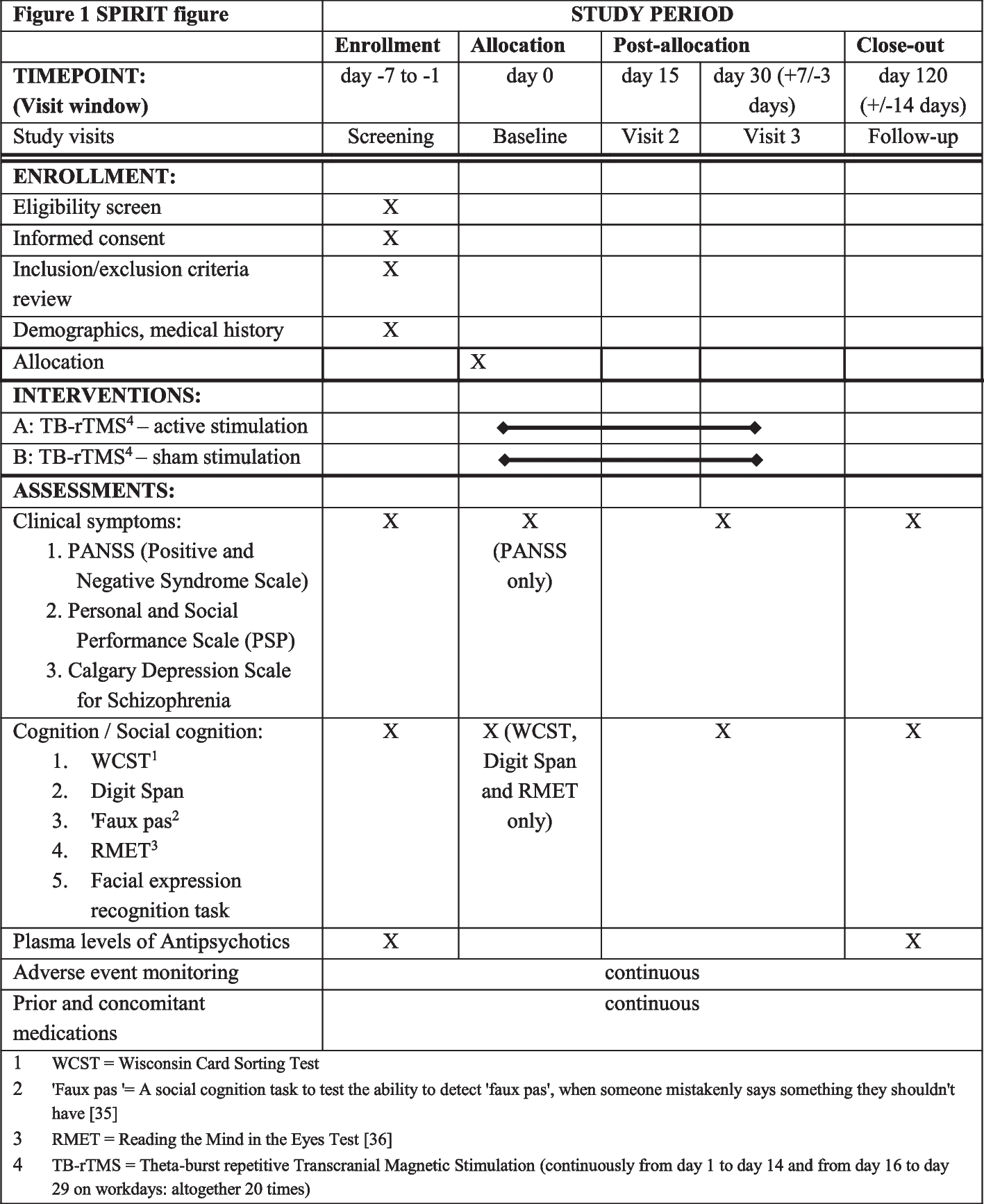

A two-group parallel, single-centre, superiority, randomised controlled trial (RCT) with an allocation ratio of 1:1 will be conducted and reported in accordance with the CONSORT guidelines [27]. A total of 150 patients undergoing elective surgery will be randomised to the control (standard preoperative care) or intervention (standard preoperative care plus VR intervention) groups. The intervention will be delivered using the Oculus Quest 2 Virtual Reality Headset™ fitted with a smartphone (Fig. 1). This device has been chosen because it is the most advanced commercially available all-in-one VR device from Oculus, equipped with a highly immersive headset, two controllers, and integrated headphones. This study will be conducted in the surgical unit of a major Australian metropolitan hospital.
Fig. 1
Oculus Quest 2 Virtual Reality Headset™
Study aimThe aim of this study is to evaluate the effectiveness of using VR technology for perioperative anxiety among adults undergoing elective surgery.
HypothesisCompared with standard care, a VR intervention delivered during the preoperative period will significantly reduce perioperative anxiety in adult elective surgical patients by 20 points on a 100-point visual analogue scale for anxiety (VAS-A).
RecruitmentThe primary investigator will consult the hospital’s admission team to review the surgical list and identify potential participants. The potential participants will be asked to participate in the study via the hospital confirmation SMS providing them details of their surgery. They will be provided with a participant information statement and consent form via a link in the SMS. Potential participants will be requested to return their signed consent prior to their day of surgery if they are interested in participating in the study.
Inclusion and exclusion criteriaPatients will be eligible for inclusion if they are 18 years old and over, scheduled for elective surgery, can understand English, and can follow instructions. Patients will be excluded if they are scheduled for day surgery, have a history of seizures or motion sickness, have visual impairment, or are unable to complete a self-reported questionnaire.
ScreeningPatients who meet the above inclusion criteria will be screened for their anxiety level using a validated tool—the Amsterdam Preoperative Anxiety and Information score (APAIS) [28, 29]—on the day of their surgery in the preoperative holding area. Patients with moderate to high preoperative anxiety levels—described by preoperative anxiety score ≥11 [30]—will be included in the study and randomised to the intervention or control group.
The APAIS is a rapid and clinically practical assessment tool developed by Nelly Moerman and previously validated against other scales to evaluate patients’ preoperative anxiety with good sensitivity and strong specificity for clinically significant anxiety [28, 29]. It consists of six questions in total. Each question is rated on a 5-point Likert scale from “not at all” to “extremely”. The sum of scores from questions 1, 2, 4, and 5 show the anxiety level, while the sum of scores from questions 3 and 6 show the level of information required by each individual. A patient with a score of 11 or more (scoring range from 4 to 20) on the anxiety scale experiences anxiety and requires further intervention [28, 29, 31]. On the need for information scale, patients scoring 2–4 are categorised as having minimal or no need for information, patients scoring 5–7 are categorised as having an average need for information, and patients scoring 8–10 are considered as having high need for information [28, 31].
Randomisation and allocation concealmentParticipants will be randomly allocated to intervention or control via block randomisation on a 1:1 ratio (control: intervention) using a computer-generated random number sequence created by an independent statistician. In addition, randomisation will be stratified by surgery type (i.e. ENT surgery, general surgery, neurosurgery, maxillofacial surgery, urology surgery, vascular surgery, orthopaedics, and other surgeries). Electronic allocation via an online platform (the REDCap™ randomisation module) will be used to conceal the treatment allocation from the researcher until they have been deemed eligible and screened.
BlindingThe Research Assistant involved in outcome data collection will be blinded to group allocation, as will the statistician who will conduct the data analysis. Clinicians (nurses, doctors, and other health providers) will not be told who is participating in the study nor their group allocation; however, it is acknowledged that it is not possible to prevent participants from sharing this information with them.
Intervention and controlThe participants will be asked to wear the VR headset for 10 min, which has been suggested in the literature to be the optimal time frame for VR without adverse side effects [32, 33]. The participants will be seated in the preoperative holding area while immersed in a VR. The preoperative holding area has been selected for administration of the intervention because evidence suggests preoperative anxiety levels among adult surgical patients peak in the preoperative holding area [34]. The VR will be used for relaxation purposes with nine natural scenes combined with natural sounds and music, for the participant to select from: blue moon, red savanna, blue ocean, green meadows, black beginning, white winter, blue deep, red fall, and the orange sunset (Fig. 2). The participant’s face and forehead will be cleaned using skin-friendly antibacterial cleaning wipes before using the VR device. Disposable hygiene covers will be used to protect the VR device. The researcher will provide the intervention and monitor the participant for adverse side effects such as headache, nausea, vomiting, sweating, fatigue, drowsiness, disorientation, and apathy throughout the session [33].
Fig. 2 Criteria for discontinuing the trial in an individual
Criteria for discontinuing the trial in an individual The researchers will monitor the participants continuously for side effects. If a side effect occurs, the VR session will be ceased immediately, and a treating medical team will be consulted. Also, there may be a risk of exacerbating the participant’s anxiety level associated with using a survey to assess anxiety. If this is the case, participants to be instructed to stop the survey and appropriate support will be provided by the researcher or medical team.
Strategies to improve adherence to interventionsThis study includes one-time intervention during the study and is a pragmatic study. The researcher will provide the intervention and the research assistant will collect the data. Researchers will daily check the data in the REDCap™ platform. Also, all researchers share information through periodic meetings (weekly) and discuss the study and appropriate management when problems occur.
Concomitant care and interventions that are permitted or prohibited during the trialThere is no care or intervention prohibited during the trial. But we anticipate that some participants may regularly use anti-anxiety/anti-depressant medications. If this is the case, the research assistant will document the name and dose of the medication taken prior surgery.
Outcomes Primary outcomePerioperative anxiety will be assessed by the Research Assistant at three time points: on admission to the preoperative area (T1), immediately before surgery (T2), and 1 h on arrival to the post-anaesthesia care unit (T3). Participants will be asked to rate their level of anxiety using the visual analogue scale for anxiety (VAS-A). The VAS-A is valid, reliable, and frequently used to evaluate perioperative anxiety levels in surgical populations [35]. The VAS-A consists of a 100-mm horizontal line representing two behavioural extremes at either end of the continuum (i.e., ‘not at all anxious’ = 0, whereas ‘extremely anxious’ = 100). Scores on the VAS-A of 25 or higher reflect significant levels of anxiety [35].

Visual analog scale for anxiety (VAS-A)
Secondary outcomesThe secondary outcomes to be assessed include:
1.Stress levels will be assessed via two methods:
A)Saliva cortisol levels as measured by collecting 4–5 ml of saliva in a clean glass tube. The samples will be labelled with participant ID number and stored at −18°C until they are transferred to Saliva and Liquid Biopsy Translational Laboratory, Queensland University of Technology, for analysis using the Cortisol ELISA Assay Kit™ by an independent laboratory. The analysis results will be intended to determine the quantitative of cortisol by an enzyme immunoassay in human saliva. This is a highly sensitive test, with results changing in minutes after exposure to an anxiety-inducing or relaxing stimulus [26]. The sample will be used for the purposes of this research project only and will be destroyed at the end of this project.
Saliva samples will be collected from participants by the Research Assistant at two-time points: on admission to the preoperative area (T1) and immediately before surgery (T2).
B)Heart rate (HR) as measured manually for 1 min and the mean HR will be calculated. The Research Assistant will record HR at three time points: on admission to the preoperative area (T1), immediately before surgery (T2), and 1 h on arrival to the post-anaesthesia care unit (T3).
2.Pain level as measured by the visual analogue scale for pain (VAS-P). The Research Assistant will ask the participants to rate their pain on the VAS-P tool at two time points: on admission to the preoperative area (T1) and 1 h on arrival to the post-anaesthesia care unit (T3).
3.Patient satisfaction as measured by the Leiden Perioperative Patient Satisfaction questionnaire (LPPSq). The questionnaire is a valid and reliable tool used to assess and measure different aspects of patient satisfaction with perioperative care with 39 questions [36]. The participants will be asked to complete the questionnaire 24 h after surgery before discharge (T4).
4.Hospital length of stay (LOS) as measured from the date of admission to discharge from hospital. The Research Assistant will collect this data from administrative records post-discharge.
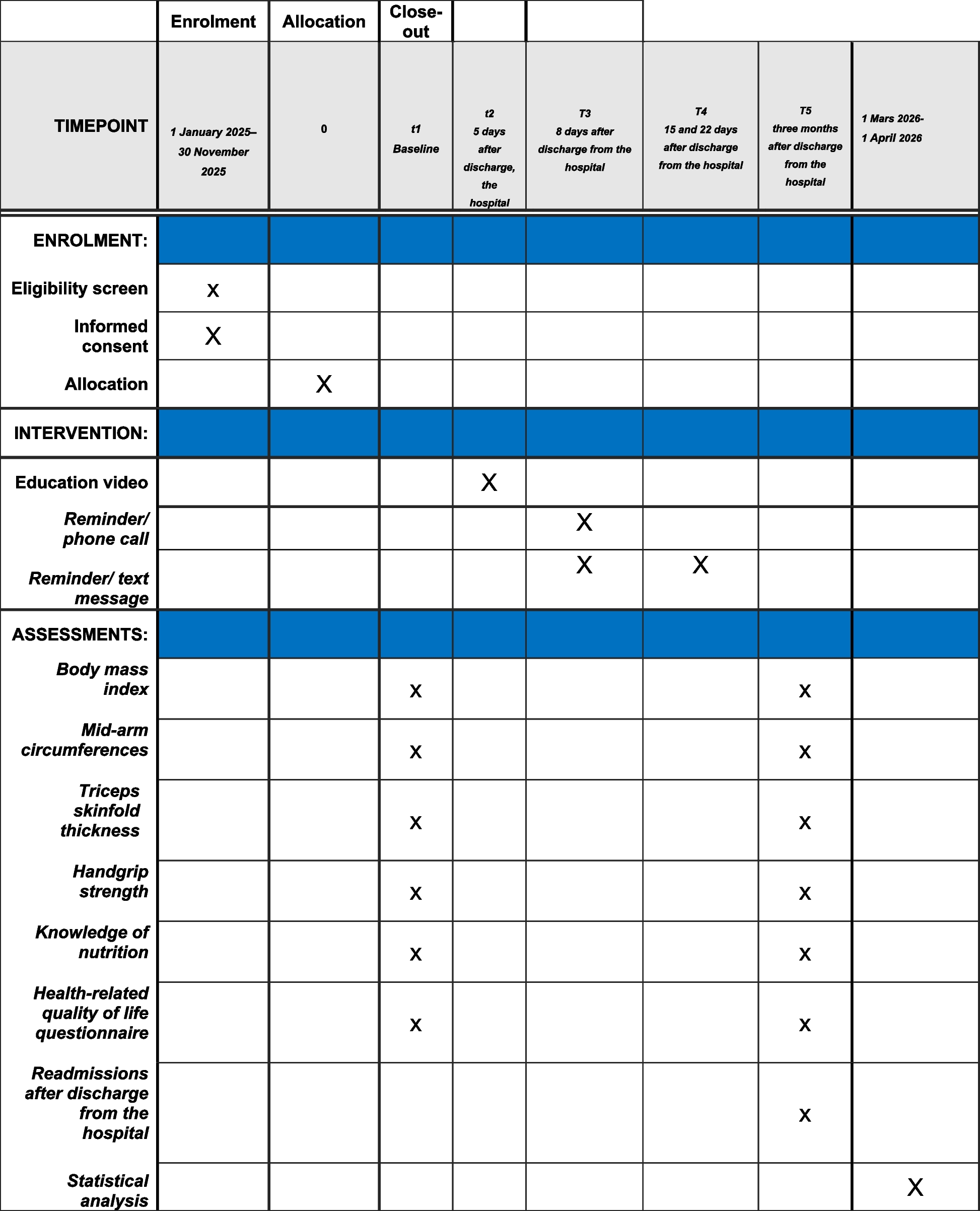
5.Adverse effects of the VR intervention at any time point as measured by the Virtual Reality Symptom Questionnaire (VRSQ). The questionnaire is a valid and reliable measure used to evaluate the possible occurrence of cybersickness symptoms, a type of motion sickness caused by exposure to VR [37]. The researcher will ask the participants in the intervention group to complete the questionnaire immediately before surgery (T2) (Fig. 3, Table 1).
Fig. 3 Table 1 SPIRIT flow diagram
Other data
Table 1 SPIRIT flow diagram
Other dataOther data will include age, sex, surgery type, type and dose of anaesthetic, anti-anxiety medications, history of previous surgeries, and discharge data. This data will be collected from the participant’s health record by the researcher.
Sample sizeBased on the previous study by Bekelis et al. [24], a sample of 150 participants (75 per group) will provide 80% power to detect a 20% change on the 100-point VAS-A between groups with a type 1 error rate of 5%. This calculation assumes a dropout rate of 15%.
Statistical planCharacteristics of groups will be summarised using counts and percentages for categorical variables and means and standard deviations for continuous variables. The primary outcome (anxiety at Time 2 and Time 3 in stacked format) will be compared between groups at each follow-up time point using a linear mixed-effects regression model, with fixed effects including treatment arm time (categorical), the interaction between treatment and time, anxiety time point (either Time 2 or Time 3), surgical type (stratification variable), and the baseline value of the outcome variable. Differences in mean VAS-A scores between groups at each time point will be presented together with 95% confidence intervals and p-values. Continuous secondary outcomes with repeated measurements will be compared using the same model. Hospital length of stay will be compared using the rank-sum test. Patient satisfaction will be compared using an independent sample t-test and descriptive statistics will be used to summarise the VRSQ.
Missing dataMissing data will be handled by complete case analysis. Data collection will occur within the same day and thus missing data should be rare.
Ethical issuesThe study, including the participant information and consent form, has been reviewed and approved by the Royal Brisbane & Women’s Hospital Human Research Ethics Committee (RBWH HREC) (HREC/2021/QRBW/74417). Participation is voluntary and decline to participate will involve no penalty or loss of benefits to which the participant is otherwise entitled, and participants may discontinue participation at any time without comment or penalty.
There are possible risks (side effects) associated with the VR intervention (38). Although rare, side effects may include headache, nausea, vomiting, sweating, fatigue, drowsiness, disorientation, and apathy [33]. Any adverse events that arise, whether related to the study intervention or not, will in the first instance be reported to the clinical nurse manager and to the researchers, who will then submit a report of the event to the HREC.
Data storage, access, and disposalUnique project codes will be assigned to participants on entry to the project and stored securely and separately from participant data. During each data collection, research staff will enter participant data into a database. At no time will identifiable study information be reported or made available to persons other than the research personnel.
All electronic data will be stored on a password-protected computer. All paper records will be kept in a locked cabinet in the Principal Investigator’s office. All data will be kept for 15 years following the project’s completion, after which time it will be destroyed or erased from the nominated drive. Only the study researchers will have access to any identifiable data, except where required by law. Data will be reviewed weekly by the researcher for auditing trial conduct.
If the participant decides to withdraw from the project, the researcher will discuss any special requirements linked to withdrawing.
Plans for communicating significant protocol amendments to relevant partiesWhen the trial protocol needs to be amended, the main investigators will discuss, communicate, and conclude the revisions. A revised protocol will also be submitted to the Ethics Committee for approval.
Dissemination plansThe results of the study will be disseminated to interested parties through publications in an appropriate journal.
留言 (0)