
記住我
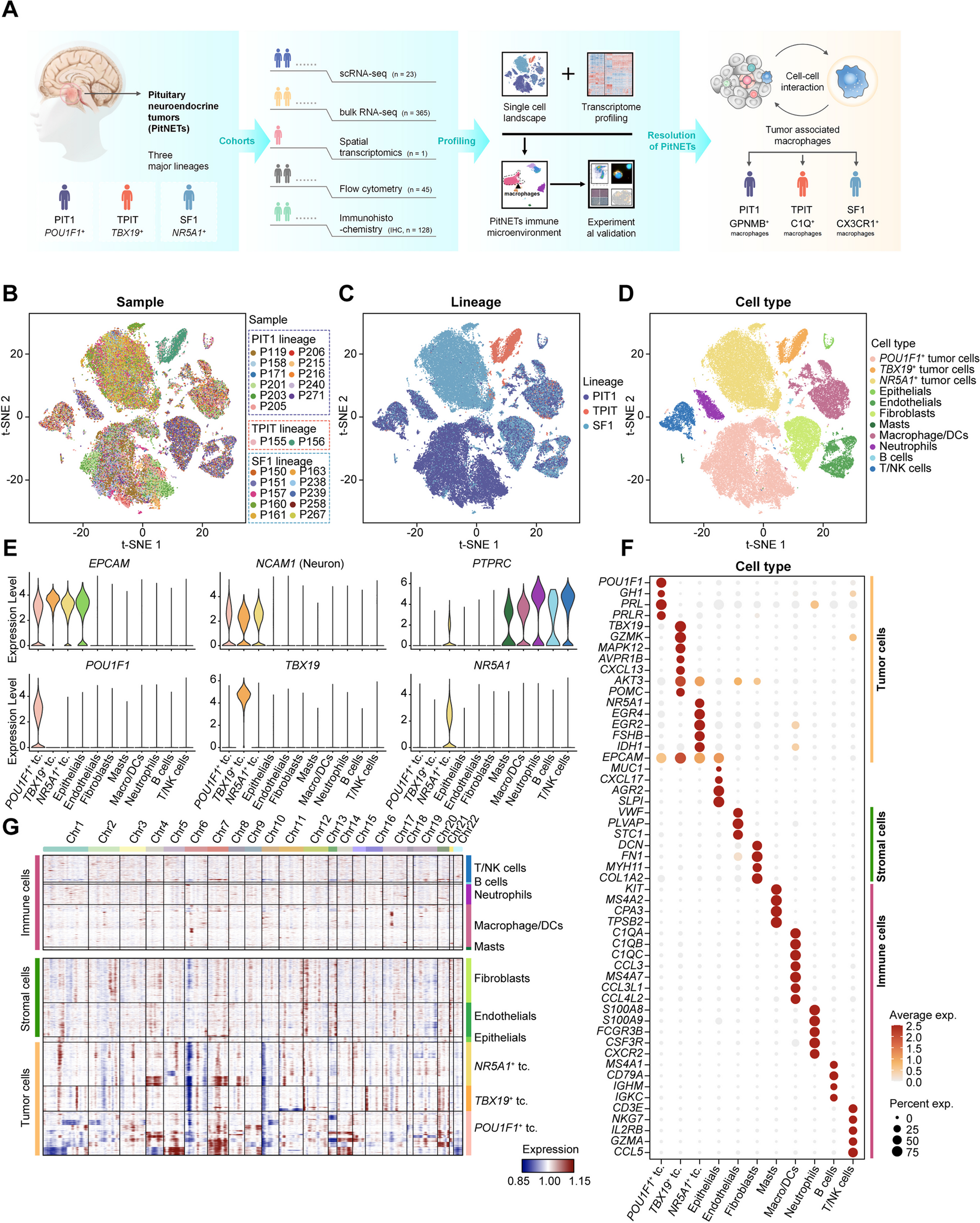
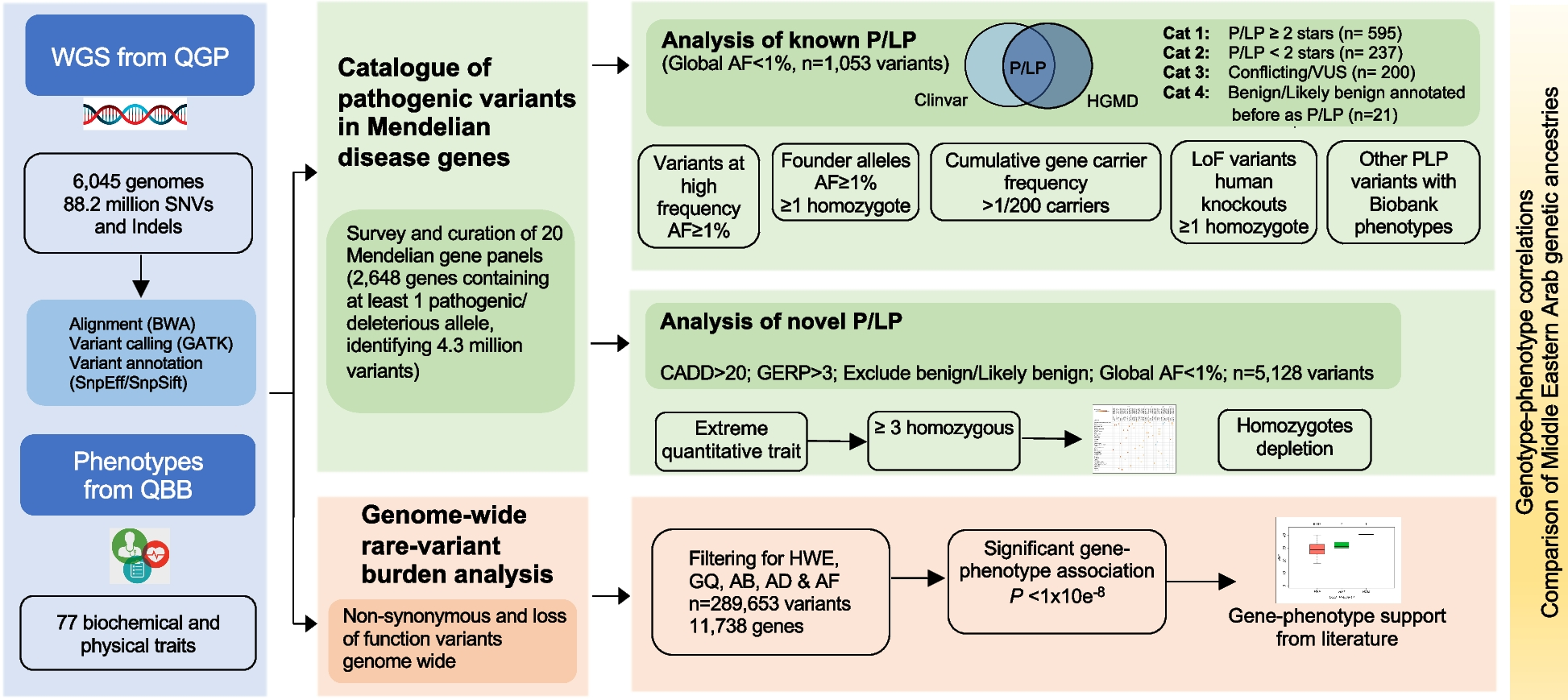
To delineate the molecular signatures of therapy response to TNF antagonists, we performed a longitudinal analysis of the blood transcriptome and epigenome of two individual clinical cohorts of IBD patients in a case-only design (Fig. 1A). For the discovery cohort (Fig. 1B), whole blood samples were collected from 14 IBD patients (10 UC/4 CD) (Table 1). Seventeen IBD patients (10 UC/7 CD), who received first-time therapy with vedolizumab [22, 23], a monoclonal antibody directed against α4β7 integrin, were used as treatment controls. For the discovery cohort, we employed RNA-sequencing on samples collected at all time points (baseline; 4, 24, and 72 h; 2, 6, and 14 weeks after the first infusion) [23]. Genome-wide DNA methylation profiling was done on baseline, week 2, and week 6 samples [33]. Therapy outcome (primary endpoint) was defined based on the achievement of clinical remission at week 14, as assessed by clinical disease activity markers (Harvey-Bradshaw Index ≤ 4 for CD; partial Mayo score ≤ 2 for UC). Of the 14 IBD patients treated with infliximab, 7 achieved clinical remission at week 14 (50%) and 9 out of 17 IBD patients (53%) treated with vedolizumab achieved clinical remission at week 14. Results from the discovery cohort were replicated in a second independent replication cohort comprising 23 subsequent IBD patients (9 UC/14 CD; Fig. 1C, Table 2) treated with a TNF antagonist (22 infliximab, 1 adalimumab) [23]. Here, samples were taken at baseline, week 2, and week 6. In this cohort, remission at week 14 was achieved in 11 patients (48%).
Dynamics of transcriptomic changes upon TNF antagonist exposureTo investigate the dynamics of transcriptional responses of IBD patients after therapeutic exposure to a TNF antagonist, we analyzed longitudinal whole blood transcriptomic data before (baseline) and at up to 6 time points after the introduction of infliximab therapy in the discovery cohort (Fig. 1A). We first compared the transcriptional signatures between remitters and non-remitters at baseline to identify any prior signature of therapy response. Through principal component analysis (PCA), we observed a suggestive ex ante separation between patients achieving remission and non-remission at week 14 along the PC2 axis (Spearman’s rho = 0.58, p-value = 0.04; Additional file 2: Fig. S1A). However, after taking the diagnosis into account, we observed that separation on PC2 mainly reflected the difference between CD and UC patients (partial correlation coefficient with diagnosis = 0.65, p-value = 0.02, partial correlation coefficient with disease status at week 14 = 0.46, p-value = 0.1; Additional file 2: Fig. S1A). The first two principal components did not associate with age, gender, or concomitant medication usage (Additional file 2: Fig. S1B, C). Differential expression analysis, after taking diagnosis as a covariate, further identified 387 genes that were nominally differentially expressed between remitters and non-remitters at baseline (Additional file 2: Fig. S1D). We next performed pairwise differential expression analyses between baseline and each of the time points after therapy initiation in the discovery cohort in remitters and non-remitters separately (Fig. 2A). Overall, treatment with infliximab led to profound alterations in the blood transcriptome within the first 24 h after drug exposure with transcript levels of most differentially expressed genes (DEGs) being downregulated (Fig. 2B, D, Additional file 1: Table S2, S3). Furthermore, we observed that patients who attained remission showed overall higher numbers of DEGs, pointing towards molecular response trajectories starting as early as 4 h after therapy exposure (Fig. 2B). The inter-individual heterogeneity, quantified by the variance in gene expression, was also significantly higher in non-remitters compared to remitters at all time points except week 14 (data not shown). ImpulseDE2 was employed to construct a continuous temporal model of gene expression [29] over time. We identified 3043 DEGs with significant impulse-like progression patterns across time points in remitting patients, whereas only 389 DEGs were identified in non-remitting patients (Fig. 2B). Pairwise and longitudinal analyses were combined in remission and non-remission patients (Additional file 1: Table S2, S3). A total of 1600 genes were shared between the groups (Fig. 2C).
Fig. 2
Dynamic changes in transcription in response to therapy induction and remission. A Schematic workflow. B Number of upregulated (dark) and downregulated (light) genes in remission (green) and non-remission (blue) patients at each time point after therapy induction obtained from the pairwise analysis and number of transiently differentially expressed genes obtained from the longitudinal analysis of the discovery cohort. Negative numbers are used to show the number of downregulated genes. C Venn diagram showing the number of DEGs in remission and non-remission patients from pairwise and longitudinal analysis combined. D Heatmap of top DEGs in remission patients from pairwise and longitudinal analysis, showing scaled mean expression counts at each time point in remission and non-remission samples. Selected immune-relevant transcripts are labeled by gene name. E Bar plot showing the number of genes in each co-expression module along with a correlation heatmap showing Spearman’s rank correlation coefficients between gene co-expression modules (columns) and clinical parameters (rows). *p-value < 0.05, **p-value < 0.01, and ***p-value < 0.001 in Spearman’s correlation. Color intensity corresponds to the correlation coefficient. F Heatmap showing Zsummary scores of baseline co-expression modules in remission and non-remission samples at weeks 2 and 6. G GO terms enriched in differentially preserved co-expression modules between remission and non-remission. Dot size is proportional to the gene ratio and color corresponds to the p-value of enrichment
Gene ontology enrichment analysis on DEGs at each time point identified complex inflammatory processes from downregulated gene sets from week 2 onwards in both remitters and non-remitters. “Positive regulation of NF-kB transcription factor activity” and “toll-like receptor signaling pathway” were among the uniquely enriched terms in downregulated genes at week 2, 6, or 14 in patients achieving remission at week 14 (Additional file 2: Fig. S2A). Terms such as “positive regulation of leukocyte degranulation” and “integrin-mediated signaling pathway” were uniquely enriched in downregulated genes in non-remission patients from 2 to 14 weeks after therapy induction (Additional file 2: Fig. S2B). Interestingly, the small set of upregulated transcripts in the non-remitter group comprised TH2- and eosinophil-related genes including ALOX15, FCER1A, and OLIG2 (Additional file 1: Table S3). These observations indicated that modulation of immune network states by antagonizing TNF in blood is complex and that even in patients who did not achieve remission at week 14, several pro-inflammatory processes are dampened in this compartment. Since classical rank-based gene expression analysis did not clearly distinguish between the therapy outcomes, we applied higher-order gene expression regulation analysis to find distinct features associated with therapy response.
Dysregulation of co-expression networks during the induction of remissionTo further annotate and condense functional groups of genes, which are linked to effective anti-TNF therapy, we next analyzed gene co-expression networks using weighted gene co-expression network analysis (WGCNA) [30]. Co-expression analysis follows the assumption that clusters of genes with similar expression patterns (so-called modules) are likely to share regulatory inputs and biological function or are derived from a specific cell type in complex tissue samples [21]. We hypothesized that co-expression patterns of differentially expressed genes could change over the course of a targeted therapy, which — although not completely unbiased — may allow a focused view on pathways that are disrupted during the induction of remission. We therefore constructed focused co-expression networks (as described in [39, 40]) using all DEGs separating remitters from non-remitters (pairwise and longitudinal combined; 3889 genes), which were identified in the previous analysis step. We started from baseline samples and compared the preservation of modules at week 2 and week 6 between patient groups, stratified according to the respective therapeutic outcome (Fig. 2A). These time points were chosen because they reflect the intermediate state between active disease at baseline and primary endpoint at which the therapy outcome was defined. This approach resulted in a total of 24 co-expression modules (Fig. 2E, M1–M24). We calculated the respective eigengene values, which represent a single expression profile for all genes within a module and correlated these values to respective clinical parameters (Fig. 2E) as well as to computationally inferred cell type proportions (Additional file 2: Fig. S2C) [41].
We next analyzed the preservation of modules at week 2 and week 6, stratified according to the respective therapeutic outcome at week 14 (Fig. 2A). We applied Zsummary statistics as a measure of module preservation [32]. M6, M7, M8, M14, and M16 modules were moderately preserved in non-remission (2 < Zsummary < 10) while not preserved in remission (Zsummary < 2) at one or both time points (Fig. 2F, G, Additional file 2: Fig. S2D). Modules M12 and M19, on the other hand, were highly preserved in non-remission (Zsummary > 10) and significantly less preserved in remission (2 < Zsummary < 10) (Fig. 2F, Additional File 2: Fig. S2D). Genes in the differentially preserved modules were involved in diverse biological processes, e.g., type I interferon signaling pathway, MDA-5 signaling pathway and interleukin-1 beta secretion in module M7 and platelet aggregation, erythrocyte development, and ROS signaling in M12 (Fig. 2G). Altogether, using co-expression analysis, we identified salient transcriptional modules that change during the therapy, specifically in patients that achieve remission at week 14.
Unique molecular signatures induced by TNF inhibitionTo identify the unique molecular signatures induced by treatment with infliximab, we compared the differentially expressed genes at each time point in IBD patients treated with infliximab to that in IBD patients treated with vedolizumab who attained remission after 14 weeks of respective therapy initiation. We found that the transcriptional dysregulation observed within the first 24 h in infliximab-treated patients was not shared in vedolizumab-treated patients (Fig. 3A). We observed three large groups of overlapping genes between the two treatments: (1) downregulated genes at early time points (4h, 24h) in infliximab-treated patients that were upregulated at later time points (weeks 2 and 6) in vedolizumab-treated patients, (2) upregulated genes at early time points (4h, 24h) in infliximab-treated patients that were downregulated at later time points (weeks 2, 6, and 14) in vedolizumab-treated patients, and (3) shared downregulated genes in both treatments at later time points (Fig. 3A). The first two groups that showed contrasting expression patterns between the two treatments could describe the unique mechanism of action of infliximab while the third group could represent the overall signature of healing and decline in inflammation. Group 1 genes were enriched in processes related to transcription and splicing as well as V(D) J recombination and mainly consisted of genes that were highly expressed at baseline in infliximab-treated patients (Fig. 3B, C). Group 2 genes were related to complement activation, leukocyte migration, and endocytosis and showed a strong upregulation at 24h specifically in patients remitting after 14 weeks of infliximab treatment (Fig. 3B, C). The last group (group 3) had a similar expression pattern at later time points between the two treatments and consisted of genes related to neutrophil degranulation and humoral response (Fig. 3B, C). Taken together, we identified a transcript signature that was regulated in a contrasting manner between treatments that target TNF and α4β7 integrin as well as genes that indicate a systemic reduction in inflammation that were shared between the two treatments.
Fig. 3
Comparison of transcriptomic changes between infliximab and vedolizumab patients. A Cross-tabulation of genes differentially expressed in patients treated with infliximab (rows) and vedolizumab (columns) that achieved remission after 14 weeks of the respective therapy induction. The three groups of overlapping genes are highlighted in orange (group 1), green (group 2), and blue (group 3). B GO terms enriched in genes belonging to the three overlap groups. Dot size is proportional to the gene ratio and color corresponds to the p-value of enrichment. The top five GO terms in each group are visualized. C Heatmap showing average scaled mean expression counts at each time point of selected genes in the three overlap groups
Dynamic changes in genome-wide methylationDNA methylation (DNAm) is an important epigenetic mechanism for long-term regulation of gene expression, which has been shown to be involved in the etiopathogenesis of IBD [42,43,44]. We thus analyzed DNAm signatures by bead arrays covering > 850,000 CpG sites across the entire genome before and 2 and 6 weeks after the administration of infliximab therapy (Fig. 4A). We used a pairwise approach to interrogate differentially methylated sites and regions between baseline and week 2 samples and baseline and week 6 samples [34]. We identified a total of 85,728 and 58,347 differentially methylated positions (DMPs) in remitters and non-remitters, respectively (Fig. 4B, C, Additional file 2: Fig. S3B, S3C). In the samples of patients achieving remission at week 14, a preponderance of hypermethylated DMPs was observed, constituting around 70% (30,132) at 2 weeks and 60% (43,478) of the DMPs at 6 weeks (Fig. 4B). Cellular deconvolution analysis [34] identified that major parts of the observed DNAm signatures originated from granulocytes, B cells, CD4+ T cells, and monocytes, similar to the transcriptional signatures (Additional file 2: Fig. S3A). The inferred granulocyte proportions in blood significantly decreased across time points only in remitting patients (linear mixed model ANOVA p-value = 0.043).
Fig. 4
DNA methylation analysis and integration of omics layers. A Schematic workflow. B Number of hypermethylated (dark) and hypomethylated (light) positions in remission (green) and non-remission (blue) patients at each time point after therapy induction obtained from the pairwise analysis of the discovery cohort. Negative numbers are used to show the number of hypomethylated positions. C Venn diagram showing the number of DMPs in remission and non-remission patients. D Heatmap of DMPs, which are correlated with DEGs, showing scaled mean methylation intensities at each time point in remission and non-remission samples. E Heatmap showing significant enrichment, quantified by odds ratio, of transcription factor binding sites (TFBS) in DMPs that are correlated with DEGs. Selected top TFs are visualized. F Over-representation and under-representation of DNAm-linked DEGs in co-expression modules. The over-/under-representation is quantified as the ratio of the observed and expected number of correlated genes present in each module under the chi-square distribution. G GO terms enriched in DNAm-linked co-expression modules. Dot size is proportional to the gene ratio and color corresponds to the p-value of enrichment
In total, 357 differentially methylated regions (DMRs) such as promoters, genes, CpG island, and enhancers were observed in remitters and 1163 DMRs in non-remitters (Additional file 2: Fig. S3D, S3E). The majority of the DMRs belonged to enhancer regions (348 in remission and 1147 in non-remission), consistent with the distribution of the DMPs (Additional file 2: Fig. S3B, S3D, S3E). These DMRs overlapped with binding sites for several transcription factors including IRF4, BATF, MEF2C, and MEF2A for hypermethylated regions and CEBPD and STAT3 for hypomethylated regions (Additional file 2: Fig. S3F, S3G). Interestingly, most DMPs and DMRs in non-remitting patients were transiently observed only at week 2, while many DMPs were stably regulated at week 2 and at week 6 in remitting patients (Additional file 2: Fig. S3C).
Analysis of DNAm-linked transcriptomic changesTo link DNA methylation changes to gene expression in cis, we performed an integrative analysis using a hierarchical approach [21], which identified DMPs located within 5kb upstream or downstream of the transcription start site of each DEG. We then calculated the correlation between gene expression of each DEG and methylation intensity of the corresponding DMPs (Fig. 4A). Out of a total of 85,728 remission-associated DMPs (DMPs at week 2 and week 6 combined), 5459 were in a 5-kb vicinity of at least one DEG. In total, 1253 DMP-DEG pairs (representing 763 genes) were significantly correlated. 65.9% of cases followed a canonical negative correlation (i.e., high methylation-low expression) (Additional file 2: Fig. S4A, Additional file 1: Table S4). DMPs that correlated with the DEGs showed a persistent hypo- or hypermethylation after therapy initiation in patients achieving remission at week 14 and overlapped with binding sites for transcription factors BATF, NF-κB, JunD, STAT3, and CEBPB among others (Fig. 4D, E, Additional file 2: Fig. S4C). This pattern was, however, completely absent in non-remitting patients, supporting our hypothesis of lack of long-term epigenetic changes in patients failing anti-TNF therapy (Fig. 4D, Additional file 2: Fig. S4D). We also investigated the representation of DEG-DMP pairs in the previously defined co-expression modules. DNAm-linked expression changes were significantly overrepresented in modules M3, M4, M15, and M24 (Fig. 4F, Additional file 2: Fig. S4B). These modules were correlated with the inferred proportions of neutrophils, T cells, and NK cells in whole blood (Additional file 2: Fig. S2C). As none of these modules was found to be disrupted by anti-TNF in the prior preservation analysis, this pointed to a rather stable association of identified DMPs and DEGs, possibly reflecting cell types or general inflammatory principles, such as neutrophil proportions (Fig. 4G, Additional file 2: Fig. S2C). Taken together, we were able to identify potentially epigenetically controlled transcriptional changes related to therapy response and induction of remission by integrating DNA methylation data with transcriptomic data. Although we cannot exclude that differences in cellular composition contribute to the above-mentioned observations, several immune-related features indicate a potential long-term alteration of cellular states through this epigenetic process.
Replication of molecular signatures in an independent clinical cohortWe conducted a formal replication using the same profiling methods (RNA-sequencing and DNAm bead array) in an independent prospective cohort of 23 IBD patients starting anti-TNF treatment (Table 2). To rule out any systematic difference between discovery and replication cohorts, we contrasted the baseline transcriptome signatures from both cohorts with transcriptome signature from (i) 20 healthy individuals and (ii) 15 inactive IBD patients (4 UC/11 CD). While principal component analysis (PCA) showed a separation between healthy controls and IBD patients (Additional file 2: Fig. S5A), we did not observe a significant separation between the discovery and replication cohorts confirming the absence of potential larger batch effects between the two cohorts (Additional file 2: Fig. S5A). A large proportion of disease-related DEGs (IBD vs. healthy) was shared between the two IBD cohorts (Additional file 2: Fig. S5B). The transcriptomic variation in the baseline samples of the replication cohort, represented by the first two principal components, was not associated with disease subtype, disease status at week 14, age, gender, or medication usage (Additional file 2: Fig. S5C, S5D, S5E). Despite the similar characteristics and inclusion criteria of the two cohorts (Tables 1 and 2), we observed little overlap between DEGs or DMPs at baseline (remitters vs. non-remitters) pointing to high heterogeneity of responders before treatment initiation (Additional file 2: Fig. S5C, S6A).
Next, we aimed to confirm the longitudinal transcriptional and methylation changes observed in the discovery cohort using the replication cohort. We observed that DEGs obtained at week 2 and week 6 in the discovery cohort were similarly regulated in the replication cohort, indicated by a strong correlation between log fold changes with respect to baseline in the two cohorts (Spearman’s rho = 0.78 for remission DEGs, 0.85 for remission-only DEGs, 0.54 for non-remission DEGs and 0.42 for non-remission only DEGs) (Fig. 5A, B, Additional file 2: Fig. S6B, S6C, Additional file 1: Table S5, S6). We repeated the module preservation analysis in the replication cohort and could replicate that modules M7 and M12 were significantly less preserved in remission compared to non-remission at week 6 in this dataset (Fig. 5D, E, Additional file 2: Fig. S6D). To exactly identify the genes that are responsible for loss of module connectivity upon therapy induction, we compared the eigengene-based connectivity measure kME or the module membership score, which measures the correlation between the expression of a gene to the consensus expression of the module [30]. In both discovery and replication cohorts, M7 genes such as RSAD2, RIPK2, HERC5, IFI44, CMPK2 SAMD4A, MSLN, XAF1, DDX60, RTP4, and PARP12 showed the strongest reduction in kME in remitting patients (Additional file 2: Fig. S6E). In the M12 module, genes with a loss in connectivity in remitters compared to baseline and non-remitters included SLC4A1, ANK1, BLVRB, TAL1, IFIT1B, ACKR1, FAM210B, TSPAN5, E2F5, and GATA1 among others in the discovery as well as replication cohort (Additional file 2: Fig. S6F). We also confirmed the correlation between the expression of the DEGs and their nearby methylated sites in the replication cohort. A total of 518 out of the 763 genes were also DNAm-linked in the replication cohort with the direction of correlation preserved in 322 genes (Fig. 5C, Additional file 1: Table S4).
Fig. 5
Replication of molecular signatures. A, B Comparison of log fold change of DEGs in remission (A) and non-remission (B) patients at weeks 2 (light blue) and 6 (dark blue) between discovery and replication cohorts. C Comparison of DEG-DMP correlation between discovery and replication cohorts. Gray dots represent a significant correlation in the discovery cohort while black dots significant correlation in both cohorts. D Heatmap showing Zsummary scores of baseline co-expression modules from the discovery cohort in remission (green) and non-remission (blue) samples at weeks 2 and 6 of the replication cohort. E Comparison of Zsummary scores of differentially preserved modules in discovery cohort between remission and non-remission samples at weeks 2 (circle) and 6 (triangle) in the discovery (orange) and replication (green) cohorts
Overall, most of the observed early longitudinal molecular signatures upon anti-TNF induction therapy were reproducibly associated with clinical outcome (endpoint: remission at week 14) in a second cohort of IBD patients, whereas the lack of replication of baseline difference points to a high heterogeneity of prior immune network states, at least in peripheral blood.
Comparison of IBD subtypesDue to the small sample size of the discovery cohort, we did not perform data analysis for the IBD subphenotypes separately in each cohort. However, by combining discovery and replication cohorts, we could attain enough statistical power to analyze CD and UC samples separately. Since, the replication cohort was sampled only at baseline, week 2, and week 6, we performed the pooled analysis at only these time points. At baseline, the differential expression analysis between remitters and non-remitters identified no significant differentially expressed genes in CD patients, while 1 DEG (IGHV1) was observed in the UC patients.
In the longitudinal pairwise analysis, we observed a higher number of DEGs in CD patients compared to the UC patients (Additional file 2: Fig. S7A). Comparing these results with the DEGs obtained from the analysis of IBD samples of the discovery cohort, we observed that almost all DEGs identified in UC patients who attained remission after 14 weeks were already contained in the IBD analysis whereas the pooled analysis resulted in the identification of many DEGs that were unique to CD (Additional file 2: Fig. S7B). To identify the molecular pathways involved in therapy response that differ between CD and UC patients, we performed gene ontology enrichment analysis on the DEGs unique to CD and the DEGs that were shared between CD and UC. While downregulated genes were enriched in biological processes related to general inflammatory signaling in both diseases, analysis of upregulated genes showed that T cell-specific terms (e.g., regulation of T cell differentiation: GATA3, LAG3, and TCF7) were increased only in CD patients (Additional file 2: Fig. S7C, S7D). TH2/eosinophil signature genes (ALOX15, FECR1A, and OLIG2), identified in the non-remitter group in the IBD analysis, were upregulated in both CD and UC individually as well. Overall, disease-specific analysis recapitulated the patterns observed in IBD analysis, but also revealed certain processes unique to CD.
Prediction of remission using early molecular changesNext, we tested the ability of each layer of molecular information at early time points (baseline vs. week 2) to formally predict therapy response at week 14. For this analysis, we combined the data from discovery and replication cohorts to increase the power of the initial analysis and then validated the results in a publicly available data set [14]. To compare the predictive potential of different individual and combined omics data layers, we performed feature selection and built prediction models on molecular sets derived from transcriptomic analysis (DEGs and differentially preserved modules), methylation analysis (DMPs), and integration analysis (DNAm-DEGs and DNAm-linked modules) (Fig. 6A) using a random forest model with 10-fold cross-validation. This approach showed that models performed better when features from baseline and week 2 were combined (Fig. 6B, D). Prediction models built on CD and UC samples separately performed better than the ones using the combined set of IBD samples (Fig. 6C, E, F). As a control, we also constructed prediction models based on the clinical data of the patients which included CRP, IL-6, serum levels of tryptophan [45], and disease activity scores at baseline and week 2 and compared them to the models from the molecular datasets. Age and gender were not included here since these are stable parameters and there was no significant association observed between these factors and the therapy outcome (CD: age Wilcoxon test p-value = 0.26, gender chi-square test p-value = 0.26, UC: age Wilcoxon test p-value = 0.07, gender chi-square test p-value = 0.26). Prediction from models using the clinical data performed worse than the ones built on the molecular datasets (Fig. 6E, F). The model using the selected features from the integration of DNA methylation and gene expression (DNAm-DEGs) was the best performing model for CD with 31 features (AUC=1) (Fig. 6E). In contrast, the models based on molecular signatures from individual omics layers (DEGs: AUC=0.97, #features=259, and DMPs: AUC=0.98, #features=65) outperformed the one based on DNAm-DEGs (AUC=0.9, #features=14) for UC samples (Fig. 6F).
Fig. 6
Feature selection and validation of molecular signatures. A Schematic workflow. B, D Comparison of AUC values of the ROC curves of prediction models constructed using selected baseline (white), week 2 (blue), and combined (pink) features from DEGs, DMPs, and DNAm-DEGs using a random forest approach in IBD (B), CD, and UC (D) samples from the training cohort. C, E, F ROC curves of prediction models constructed using selected features (baseline and week 2 combined) from DEGs, DMPs, DNAm-DEGs, differentially preserved, DNAm-linked, combined modules, and clinical parameters using a random forest approach in IBD (C), CD (E), and UC (F) samples from the training cohort. G ROC curve of prediction model constructed using selected features from DNAm-DEGs in the validation cohort. H Comparison of log fold change between remitters and non-remitters at baseline (white) and week 2 (blue) (left) between training cohort and validation cohorts
As overfitting is an inherent challenge of machine learning using a single cohort approach even when using cross-validation, we next tested the prediction model built on the most discriminating set of features (DNAm-DEGs) from CD in an independent set of patients from a publicly available therapy response cohort of 20 CD patients with peripheral blood gene expression data (external validation cohort, see the “Methods” section) [14]. Our model was able to predict therapy outcome in the external validation cohort with an accuracy of 85% (Table 3) and a formal prediction model built using the DNAm-DEGs in this cohort obtained an area under the ROC curve of 0.88 (Fig. 6G). In addition, we observed significant correlations in the regulation of DNAm-DEGs across time points and therapy responses between the two cohorts (Spearman’s rho = 0.52) (Fig. 6H).
留言 (0)