
記住我
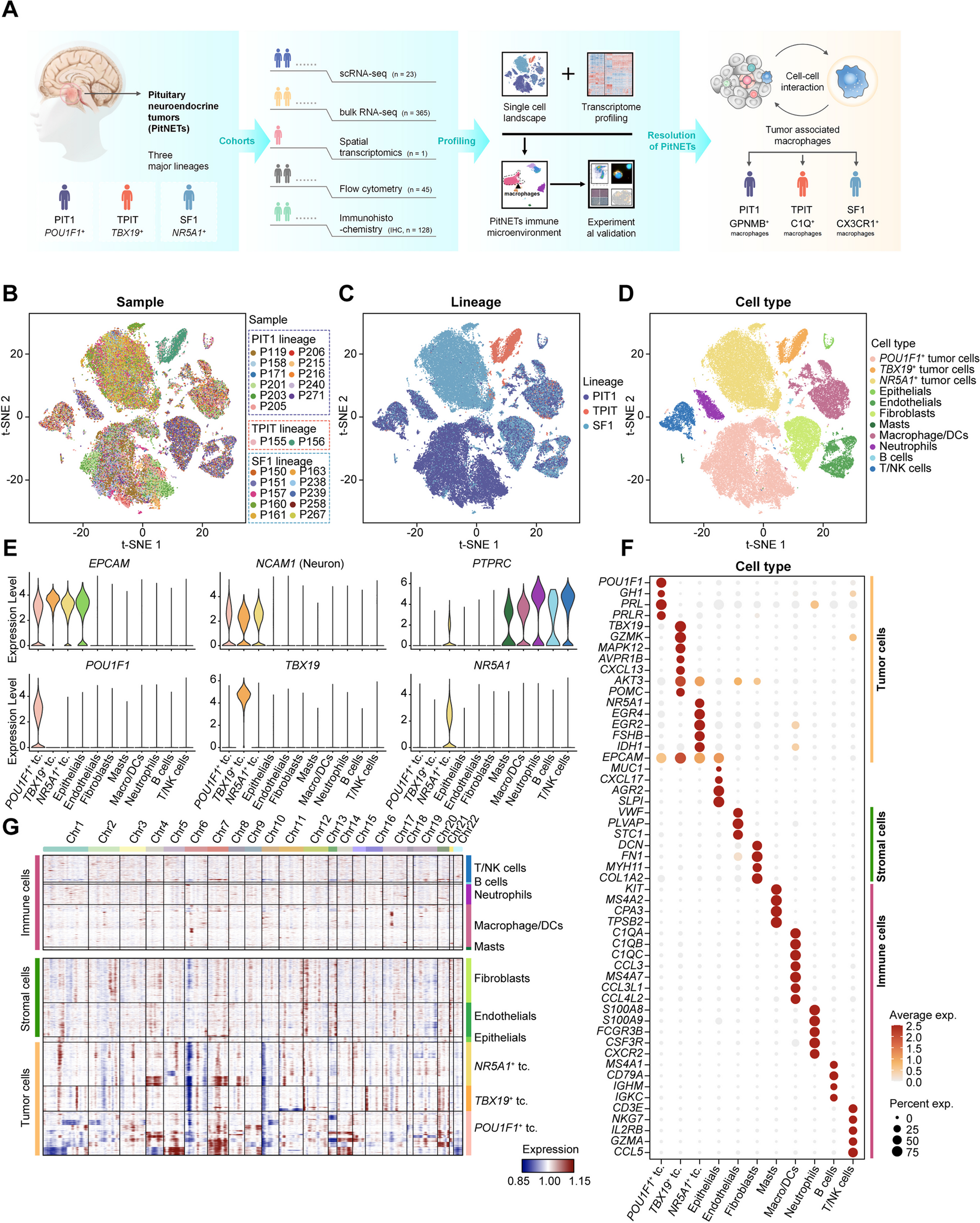
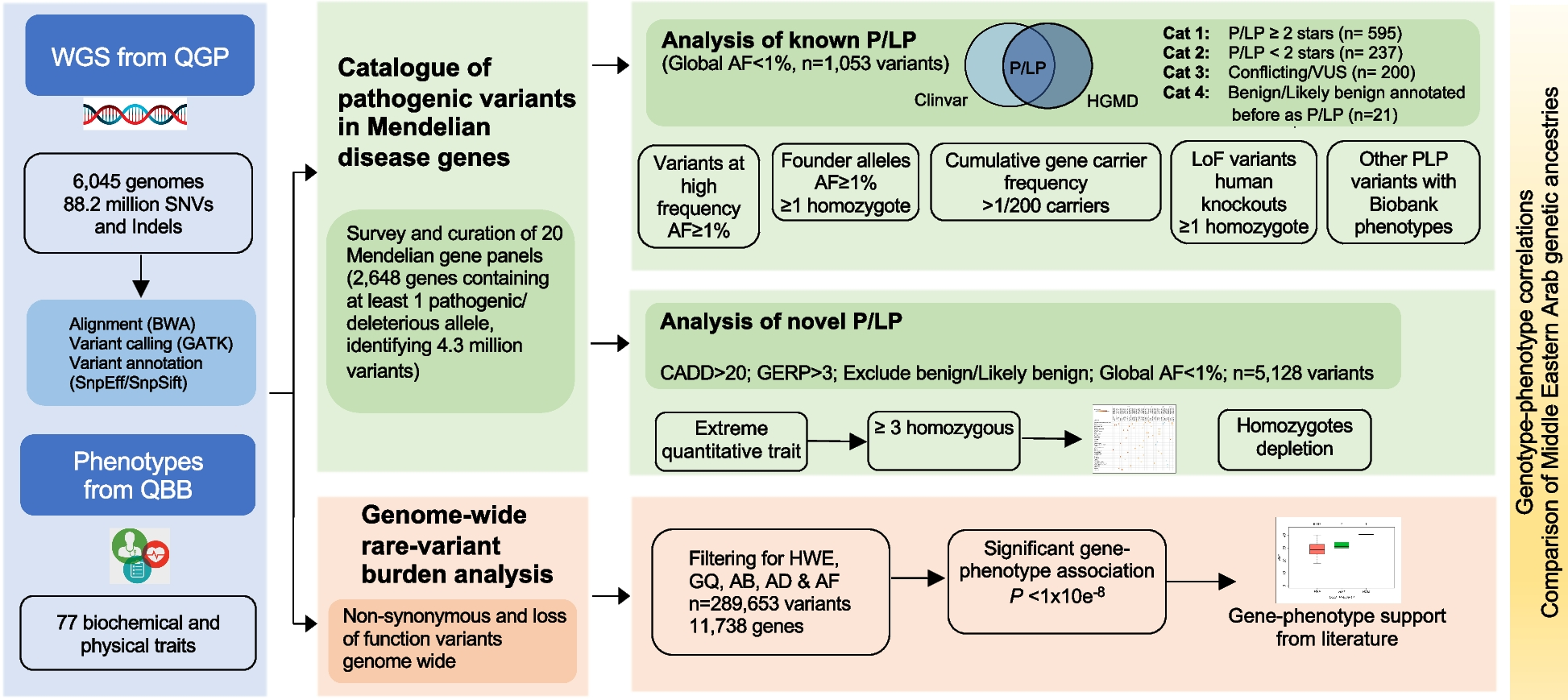
In order to systematically evaluate the contribution of recurrent genomic deletions to autosomal recessive conditions, we first mapped all possible loci that are susceptible to recurrent deletions caused by NAHR between directly oriented SDs [42, 43] using the GRCh38 human reference genome sequence (Additional file 3: Figure S1, Additional file 1: Table S2). The collapsed NAHR map contains 717 unique recurrent deletion regions. We enumerated the subset of recurrent deletion events with available data from screening efforts in the literature or clinical testing to substantiate a prevalence estimate, and focused the subsequent analyses on these genomic intervals (n=51).
We identified 30 autosomal deletions with a maximal population prevalence over 1/1,000,000 based upon estimates from the UK Biobank, the Icelandic, the gnomAD SV database, or region-specific studies [23,24,25] (Table 1, Additional file 1: Table S1). Of note, these deletion allele frequencies reflect empirical prevalence measurements from adult populations, which closely represent the effective allele frequencies (i.e., combined consideration of both the de novo mutation rate and fitness of the variant on a cellular, developmental, and organismal level) suited for recessive disease trait load estimation. These 30 deletions span 64 Mb of unique genomic sequences in the assayable portion of the human genome, contribute to an aggregate population allele burden of 1.3%, and encompass 1555 genes, of which 78 are known to cause recessive disorders. An additional 20 deletions, with populational prevalence possibly lower than 1/1,000,000, are also identified to recur in high prevalence if a clinical cohort is ascertained (Additional file 1: Table S1). With the 20 ultra-rare deletions included, the span of genomic coverage increases to 82 Mb; the number of genes involved becomes 1875, with 101 representing established recessive disease trait genes. Moreover, the “haploid genetics” concept begins to emerge as an approach based on observational data and data analyses.
Table 1 Recurrent genomic deletions that are prevalent in the populationWe then catalogued, based on existing knowledge and datasets, a compendium of all reported and predicted carrier alleles for each known recessive trait gene in the human genome. Our objective for this recessive allele catalog is to estimate and dissect the impact of the new mutation recurrent genomic deletions’ contribution to the overall disease burden. Based on mode of inheritance curations from OMIM [44], DECIPHER [45], and ClinGen [46] (data accessed on 1/4/2021), a totality of 2659 recessive disease trait genes were assembled. The carrier allele burden for each “recessive trait gene” was calculated by summing up frequencies of unique alleles for all high-quality pathogenic variants from ClinVar, all structural variants (SV) predicted to be LoF from gnomAD SV v2.1, all high-confidence LoF small variants identified in gnomAD v3.1, and the NAHR-mediated recurrent genomic deletions, if applicable. An aggregate of 85,068 small variant and large deletion carrier alleles were identified for the 2659 genes (Additional file 1: Table S3). For the 78 known recessive genes in the NAHR deletion regions, the number of per gene pathogenic alleles range from 1 to 308, with a median of 14. As a comparison, the remaining 2580 known rare recessive disease trait genes have a similar median per gene pathogenic allele count, 14, but a wider range, from 1 to 3562.
A limitation of this calculation is that SNV pathogenic missense, in-frame indel, or intronic variants not currently reported in ClinVar are inadvertently omitted. However, we argue that carrier alleles not represented in ClinVar tend to have lower allele frequencies and thus do not have a major impact on the subsequent carrier burden estimates. We further argue that the alleles that receive an entry and curation in ClinVar have higher frequencies—and therefore greater impact on recessive disease, and these are the alleles more easily ascertained in screening tests of clinical diagnostic laboratories. This latter contention is supported by the aggregate gene-level carrier allele burden from our analysis matching empirical experience in genetic testing carrier screenings results (Additional file 1: Table S4) [47].
Nevertheless, to account for potential unrepresented alleles from recessive disease trait genes that have not been scrutinized by large-scale systematic clinical or research screening, in the subsequent analyses, we supplemented the disease allele pool for each gene with a 10% extra variant load, comprised of ten hypothetical variants each accounting for 1% of the overall carrier burden (see “Methods”) for each gene. Of note, the NAHR deletion alleles rank as the most (49/78) or second most (11/78) frequent (highest population allele frequency) carrier alleles together comprising over three quarters of known recessive trait genes within NAHR regions! Even with the abovementioned conservative “padding” to represent ten hypothetical alleles not yet ascertained, the NAHR alleles still contribute to greater than 10% of the total gene-level carrier allele burden for 60 of the 78 genes (Additional file 1: Table S5).
NAHR deletions contribute a major fraction of recessive disease load to genes mapping within rearrangement hotspotsIt is important to note that, for a recessive trait, the population frequency and relativized frequency of a particular allele from a pool of alleles (Fa, fraction of allele burden) is not linearly correlated with the probability of sampling a patient with the specific allele from a group of patients (Fd, fraction of the disease burden). The distribution of alleles in affected individuals is determined by the pairwise allele frequency products in a pool.
Thus, we calculated allelic contributions to recessive disease load using an n × n Punnett square, where n is the number of carrier alleles for a recessive disease trait gene. The calculated NAHR deletion contribution to disease can be calculated from the matrix. We denote Fd as the modeled probability of sampling individual carrying at least one recurrent deletion allele from a pool of patients affected with the recessive condition caused by the same gene. We empirically considered a gene to be under significant NAHR deletion burden for population prevalence of the associated recessive disease trait, if the recurrent genomic deletion is expected in greater than 20% of all patients with this recessive disorder. By this definition, 74% (58/78) of NAHR-region recessive genes, which account for 2.184% of all known recessive genes, are under significant NAHR deletion burden for recessive disease trait prevalence (Table 2)! In the context of the other alleles from the same gene, the disease contribution of the NAHR deletion (Fd) ranks at the top for 49 genes, and at second place for 11 more genes. The Fd scores of the top 3 alleles are listed in Table 2 and Additional file 1: Table S5 to illustrate a snapshot of the allelic architecture for each recessive trait gene.
Table 2 Recessive genes with NAHR-mediated recurrent genomic deletions contributing to more than 20% of the overall disease burdenWe next defined a log-scaled index we termed the NAHR deletion’s Impact to Recessive Disease (NIRD), to depict the gene-level disease load contribution of the NAHR allele relative to an allele with a median level of contribution to the same gene among all population carrier alleles (See “Methods” section). A positive NIRD score predicts that the NAHR deletion allele plays a predominant (above the typical allele) role among all carrier alleles of the gene in disease contribution, whereas a negative score predicts a minor (below typical) role. Known recessive genes in the recurrent deletion region tend to have high NIRD scores, with 91% (71/78) scoring above 0, and 79% (62/78) scoring above 2. Of note, the two highest NIRD scores are found in RBM8A and NPHP1, 9.8 and 7.7, respectively. Both are extremely large values considering the NIRD is log-scaled.
To appreciate the properties of the NIRD scores, we adjusted the algorithm to calculate the disease contribution of any given pathogenic allele for a recessive trait gene, as an Allelic Impact to Recessive Disease (AIRD) score. The most common carrier allele observed in cystic fibrosis, NM_000492.3(CFTR):c.1521_1523delCTT (p.Phe508delPhe), also known as the ΔF508 allele, has an AIRD score of 7.3; the third most common carrier allele for Niemann-Pick disease type A, NM_000543.5(SMPD1):c.996del (p.Phe333fs), has an AIRD score of 2; a well-known founder mutation observed in ~10% of patients of Ashkenazi Jewish descent with Tay-Sachs disease, NM_000520.6(HEXA):c.1421+1G>C, has an AIRD score of −0.15, due to its lower allele frequency of 1.97 × 10−5 in the general population according to gnomAD v3.1.
The NIRD and related findings provide the computational framework that supports two consequences. First, for the ~2% of known human recessive genes genome-wide or 74% of recessive genes in NAHR regions, one of the most effective but under-utilized approaches and strategies for identifying novel disease-causing alleles from human subjects for these genes is to sequence affected individuals carrying the heterozygous recurrent genomic deletion encompassing the gene of interest. Second, there likely exist uncharacterized recessive disease trait genes that may be most effectively identified by sequencing individuals bearing prevalent recurrent genomic deletions—i.e., any of the remaining 1477 genes within these deletion regions may have yet to be assigned an AR disease trait and could be novel biallelic/recessive disease trait genes.
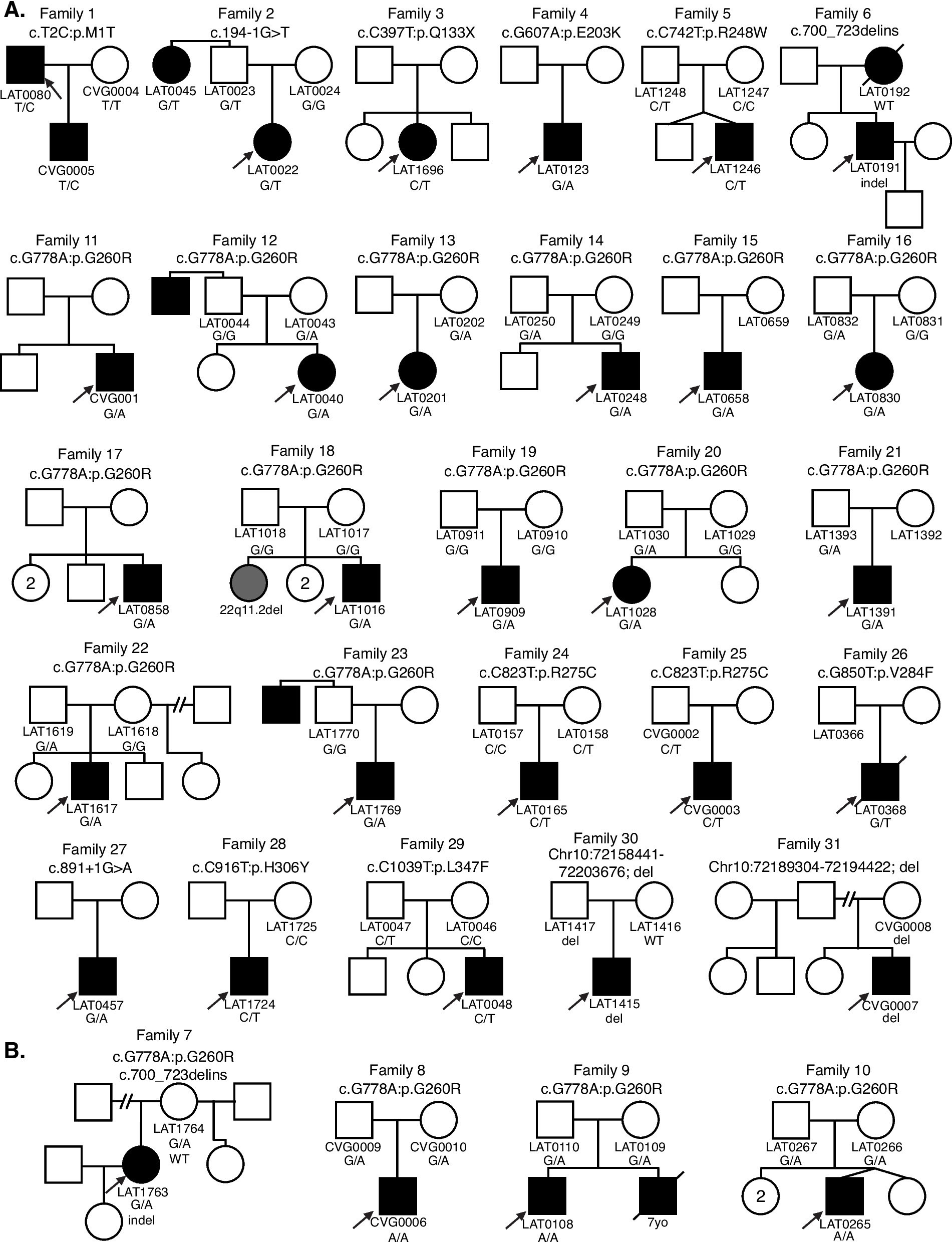
Meta-analysis suggests under-representation of the NAHR deletion alleles in currently discovered recessive disease trait allele poolsThe striking prediction of the high contribution of NAHR deletions to relevant recessive disease trait load is seemingly contradictory to our current impression of the recessive allele landscapes. This implication led us to hypothesize that the NAHR deletion alleles are currently under-represented in disease characterization efforts. To test this latter hypothesis, we analyzed the distributions of a near-complete catalogue of currently discovered disease alleles in 181 patient families affected with one of the recessive traits whose carrier burden are predicted to be almost exclusively from NAHR-mediated large deletions (Fd > 70% from Table 2). The cohorts are assembled by meta-analysis of all literature reports for patients with the corresponding recessive disease trait disorder recorded in HGMD (version 2020.4), with the assumption that most patients, penetrant for the clinical disease entity, with these extremely rare recessive disease trait disorders characterized in research efforts are reported in the literature. NPHP1, the top-ranking gene from Table 2, is a well-characterized recessive trait “disease gene,” for which many research characterized patients may not result in published literature. Therefore, NPHP1 is not included in these analyses because the literature-assembled meta-analysis cohort is unlikely to represent the natural disease allele composition (i.e., clinical practice) in the world.
It is expected that all patients with biallelic disease variants fall into three categories (1) HMZ: those affected with homozygous small variants possibly from a close- or distant- consanguineous relationship, (2) SNV+SNV: those affected with compound heterozygous small variants, and (3) NAHR deletion CNV+SNV (NAHRdelCNV+SNV): those affected with a large deletion in trans with a small variant allele. We anticipate that category #1-HMZ accounts for a substantial proportion, demonstrating the well-established robustness of autozygosity mapping as a method for allelic and new recessive trait gene discovery (as populational rare alleles can be escalated to much higher clan allele frequency) [14]. In outbred pedigrees and populations corresponding to categories #2-SNV+SNV and #3-NAHRdelCNV+SNV, our modeling from the NIRD hypothesis is that #3-NAHRdelCNV+SNV should account for a higher fraction. The opposing trend would suggest that our current disease gene/allele discovery efforts are not exploiting the large deletion allele to the fullest extent that a “human haploid genetics” approach might allow.
All the genes except for RBM8A show a poor representation for the #3-NAHRdelCNV+SNV configuration, based on the 104 families when excluding the ones affected with RBM8A variants from the entire cohort (Additional file 1: Table S6). Note that since most of the variants reported in these families are only documented once in affected human subjects, we cannot rule out the possibility that some of these variants are not causative to the clinical presentation, i.e., the variants of interest are not pathogenic determining alleles. More than two thirds (71/104) of these families carry homozygous disease alleles (57 unique alleles). Based on our modeling and the assumption of random mating, patients with homozygous variants are expected to account for a small fraction of the overall cohort, ranging from 0.53 to 5.4% per gene. However, the observed fractions of homozygotes for each gene are 1.9 to 189 (median 47) fold higher than expected. Furthermore, all of the 71 (or 57 unique) homozygous variants are rare, with 43 being ultra-rare (as defined by not observed in gnomAD v3.1). The collective patterns suggest that current efforts investigating these recessive traits tend to ascertain patients from populations with elevated autozygosity or from targeted population groups with their ethnic-specific disease founder alleles.
To avoid potential confounding factors from study designs and patient ascertainment methods, we removed patients with homozygous variants and focused on those with compound heterozygous variant alleles. Our modeling predicts that for these top-ranking genes analyzed, the number of patients with NAHRdelCNV+SNV should be 2.8 to 20 (median 10) fold higher than the number of patients with compound heterozygous small variants (Additional file 1: Table S6). The observed counts from many individual genes are too low to support a meaningful conclusion, but in aggregate, we have identified fewer patients with NAHRdelCNV+SNV [14] compared with patients with compound heterozygous small variants [16]. The recurrent deletions involved are 16p13.11 (n=4), 17p12-HNPP (n=3), DiGeorge 22q11.2 (n=2), 10q11.21q11.23 (n=2), 1q21.1-TAR (n=1), the Smith Magenis syndrome deletion (n=1), and proximal 16p11.2 (n=1). The poor representation of deletion-bearing patients shows a bias that under-represents category #3-NAHRdelCNV+SNV and deviates from the expectation driven by our analysis using empirical population allele frequencies and the NIRD score.
RBM8A is the only gene from our analysis that demonstrated a discovery pattern favoring #3-NAHRdelCNV+SNV, with the majority (95%, 73/77) of patients affected with the RBM8A- thrombocytopenia-absent radius (TAR; OMIM #274000) syndrome being compound heterozygous for the 1q21.1-TAR deletion and a small variant, whereas no patients were found to carry homozygous RBM8A pathogenic variants (Additional file 1: Table S6). This finding is consistent with expectations from our computational modeling based on the allelic spectra illustrating an overwhelming fraction of contribution of the 1q21.1 NAHR deletion at the disease locus (Tables 2 and S6). Moreover, the observed representation of NAHRdelCNV discovery at this locus, in contrast to other loci, is expected because of a unique characteristic of the RBM8A-1q21 locus. The disease presentation requires a combination of the rare 1q21 NAHR deletion null allele and a common (~1% minor allele frequency) hypomorphic small variant [48]. However, neither of the two allele types can be found in patients as homozygotes—the NAHR deletion homozygotes are lethal and the homozygous hypomorphic small variants are not disease-triggering. The unique molecular allele architecture and disease pathogenic mechanism of RBM8A, a condition that is clinically uniform and genetically homogeneous, shuts the door of discovery by sequencing of population with high autozygosity, but spontaneously presented the #3-NAHRdelCNV+SNV configurations for research discovery [49]. Similar expectations, empirical modeling and observations for Tbx6-derived scoliosis, i.e., TBX6-associated congenital scoliosis in mice, were found [26, 29].
A human haploid genetics and genomics approach to recessive trait genesWe retrospectively analyzed two existing clinical cohorts to find data that test our computational prediction of NAHR deletions conferring a major disease burden to many recessive disease traits. The configurations of the two cohorts are not optimized for discovery, but seem to have provided preliminary evidence in support of our prediction from computational modeling. The first cohort was assembled focusing on the COX10 gene, defects of which cause mitochondrial complex IV deficiency (OMIM# 220110) inherited as an AR trait.
COX10 is located within the 17p12 recurrent deletion that is associated with hereditary neuropathy with liability to pressure palsies (HNPP, OMIM# 162500), a mild form of peripheral neuropathy, or a dominant susceptibility locus to neuropathy after traumatic injury, akin to an animal model observed as the Wallerian degeneration slow phenotype modeled in the Wld triplication mouse [50]. HNPP is due to decreased dosage of the PMP22 gene via haploinsufficiency and is inherited as a liability to pressure palsies originally described in the Dutch population and pathologically presenting as tomaculous neuropathy [51, 52]; it is often only manifested clinically as multifocal neuropathy elicited after sustained trauma to a peripheral nerve that traverses close to the body surface and manifest as an entrapment neuropathy [53] or an operative carpal tunnel syndrome co-segregating through multiple generations [27]. PMP22 maps within the 1.5 Mb HNPP deletion CNV and COX10 is the only gene in the deletion interval with a known AR disease trait association other than PMP22; the latter PMP22 is associated with both an AD and AR neuropathy traits [54, 55]. Based on our calculation, ~77% of all patients affected with biallelic COX10 pathogenic alleles in an outbred population carry one HNPP deletion (Table 2).
We retrospectively investigated results from 596 patients suspected with a mitochondrial disorder who were clinically tested for COX10 coding region sequencing and deletion/duplication CNV analyses. The strength of the patient ascertainment strategy from this cohort is that patients were referred based on clinical suspicion, and therefore the distribution of pathogenic alleles from this cohort is likely free of a “molecular diagnosis bias.” A weakness of this cohort configuration is that the selected disease phenotype is of high genetic heterogeneity, which inherently predicts that only a small number of patients will indeed be affected with a COX10-related condition. Nevertheless, we found two patients received a possible molecular diagnostic finding in COX10, both carrying the HNPP deletion as one allele.
In subject #1, a hemizygous variant resulting in an in-frame small duplication of two amino acids, c.1277_1282dup (p.M426_L427dup) in exon 7 of COX10, was identified in trans to the HNPP deletion (Additional file 3: Figure S2). Subject #2, whose referral indication is COX deficiency, has a rare VUS c.858G>T (p.W286C) in COX10 in trans with the HNPP deletion. In the remaining patients without a definitive molecular diagnosis, two patients were found to have the heterozygous HNPP deletions, but a second hit in COX10 was not found, although we cannot rule out the possibility of additional findings in intronic or regulatory regions. These findings, though under-powered, are consistent with our prediction that most patients with cytochrome c oxidase deficiency carry one HNPP deletion allele, either de novo or inherited. Considering the high frequency of the HNPP susceptibility allele [20] with absence of selection and late-onset adult disease [56], it is possible that more novel COX10 disease alleles can be revealed by sequencing individuals with the HNPP deletion and a mitochondrial spectrum of clinical phenotypes, thereby improving our understanding of the biological function of the COX10 gene.
The second cohort we assembled is based on the criteria that a patient carries one of the NAHR deletions and that genotype information of the non-deleted allele is available for analyses. Thus, we identified such individuals from a cohort of 11,091 subjects who were referred for clinical exome sequencing (cES) at a diagnostic laboratory due to a differential clinical diagnosis including various suspected genetic disorders. We performed an initial screen for patients carrying one of the genomic deletions from Table 1, which resulted in 161 subjects carrying one recurrent deletion and 3 subjects carrying two. The two most frequently observed types of deletions, the 15q11.2 BP1−BP2 deletion (n=41) and the NPHP1-2q13 deletion (n=23), are excluded from downstream analysis. This exclusion is because none of the coding genes from the 15q11.2 BP1−BP2 deletion have been implicated to be associated with a Mendelian disease trait [57], and the critical gene at 2q13, NPHP1, has been already extensively studied [58]. We also excluded six subjects harboring the X-linked, hemizygous deletion in the Xp22.31 STS locus. After excluding these three groups of deletion CNVs, cES data from personal genomes of 95 subjects, collectively harboring 96 incidences or 26 types of recurrent genomic deletions, were available for us to build the second cohort (Additional file 1: Table S7).
This second cohort is not optimized for discovery because it is a collection of various different deletions without any enrichment for a targeted phenotype. Additionally, despite a subset of these patients carry one of the disease-associated large deletions that are known before cES, they are still referred for cES analyses; such a property predicts that the disease pathogenesis mechanism found in this cohort tend to be more complex than a typical Mendelian disease cohort. Such individuals may more likely to be represented by a “blended phenotype” [59].
Again, in accordance with our expectations, more than a quarter (26/95) of these subjects were found to have probable small variant molecular diagnostic findings independent from the deletion. From the remaining 69 subjects with an apparent undiagnostic cES result, we identified 4 subjects with rare variants in coding regions exposed by the deletion as potential molecular diagnoses (Table 3). The first patient is subject #1 described above with HNPP deletion and a COX10 small variant allele.
Table 3 Clinically significant sequence variants uncovered by the deletions. Subjects #1 and #2 were identified in a COX10-phenotype-driven cohort analysis. Subjects #1, #3, and #4 were identified in the molecular-deletion-driven clinical exome data reanalysisThe second patient, subject #3, has clinical features including ataxia, developmental delay, microcephaly, and short stature. A recurrent 10q11.21q11.23 deletion [60] was identified in trans to a novel missense variant allele c.1490T>C (p.F497S) in the ERCC6 gene. Biallelic variants in ERCC6 are associated with cerebro-oculo-facio-skeletal syndrome 1 (COFS1, MIM# 214150) or Cockayne syndrome type B (CSB, MIM# 133540). The high allele frequency of the 10q11.21q11.23 deletion (1.412×10−4) increases the probability for a second allele with ultra-low frequency, like the c.1490T>C (p.F497S) ERCC6 variant, to be correlated with a set of human clinical phenotypes.
Subject #4 presented with severe neurodevelopmental diseases and dysmorphic features. We identified a hemizygous OTUD7A frameshift variant allele c.2023_2066del (p.D675Hfs*188) in trans with the recurrent 15q13.3 BP4-BP5 deletion, providing evidence for OTUD7A as a new disease gene. The recurrent deletion mediated by BP4 and BP5 at the 15q13.3 locus is associated with highly variable NDD (neurodevelopmental disorder) phenotypes, ranging from asymptomatic to mild to moderate intellectual disability, epilepsy, behavioral issues distinct from neurotypical behaviors (e.g., autism spectrum disorders, attention deficit hyperactivity disorders), and variable dysmorphic features [61, 62]. While heterozygous deletion causes highly variable phenotypes, reported homozygous 15q13.3 BP4-BP5 deletion consistently manifest disease phenotypes including significant NDD, epilepsy, hypotonia, visual impairments, and other less common phenotypes including autism spectrum disorder, short stature, failure to thrive, microcephaly, and variable dysmorphic features (Additional file 1: Table S8) [63,64,65,66,67]. The critical gene responsible for this “ciliopathy like clinical presentation” of the 15q13.3 BP4-BP5 deletion has been debated, but evidence suggests that OTUD7A, encoding a member of a family of deubiquitinating enzymes, may be a plausible candidate [68, 69].
Studies using syntenic heterozygous deletion mouse models suggest a critical role of Otud7a in neuronal development and brain function [68, 69]. Otud7a-null mouse models manifest many cardinal features of the 15q13.3 deletion syndrome [68]. The c.2023_2066del (p.D675Hfs*188) variant identified in subject #4 maps to the last exon of the OTUD7A gene, and is thus predicted to not result in nonsense-mediated decay (NMD) [70]. However, the variant is predicted to result in substitution of the C-terminal amino acids after aspartic acid with 187 novel amino acids and a premature termination of the protein translation (PTC). This change may remove the C-terminal Zinc finger A20-type domain and abolish the normal function of the protein. Our finding in Subject #4, together with recent case reports of patients with a homozygous missense OTUD7A variant alleles [71], or compound heterozygous 15q13.3 deletion in trans with a frameshift OTUD7A variant [72], supports our contention and corroborates that OTUD7A may be the critical “driver gene” in the 15q13.3 deletion syndrome. OTUD7A may be sensitive to gene dosage effect and contribute to disease etiology at least in part through a biallelic AR disease trait mechanism.
Interestingly, we observe that the population small variant allele pool for OTUD7A is depleted for LoF alleles based on gnomAD. Without the 15q13.3 deletion contributing to a major carrier burden, the paucity of small variant disease alleles for OTUD7A would make disease association establishment using patient data much more challenging. From an alternative perspective, OTUD7A’s current apparent “high” gene intolerance to haploinsufficiency (pLI=0.95) may have incidentally portrayed it as a “dominant” Mendelian disease gene, whereas the calculated high NIRD score (5.3) of the gene strongly indicates that the intolerance to haploinsufficiency should be much lower, i.e., low likelihood of being an AD trait gene.
In Subject #5 with severe NDD, we identified a c.649dup (p.R217fs*8) pathogenic variant in the PRRT2 gene in trans with the recurrent 16p11.2 BP4-BP5 deletion, providing compelling evidence for a novel disease AR trait inheritance mechanism for PRRT2. The 16p11.2 BP4-BP5 recurrent deletion is known to be associated with mild dysmorphisms, macrocephaly, and neuropsychiatric phenotypes including DD/ID and autism spectrum disorder (ASD) with incomplete penetrance, a NDD [73, 74].
The PRRT2 gene is highly expressed in mouse brain and spinal cord during early embryonic development [75]. Heterozygous LoF variants in PRRT2 cause movement and seizure disorders including familial infantile convulsions with paroxysmal choreoathetosis (OMIM# 602066), episodic kinesigenic dyskinesia 1 (EKD1, OMIM# 128200), or benign familial infantile seizures 2 (BFIS2, OMIM# 605751), with incomplete penetrance documented [76]. The c.649dup (p.R217fs*8) allele is the most frequent pathogenic variant, occurring at a mutational hotspot with homopolymer of 9 cytosine bases adjacent to 4 guanine bases that are susceptible to DNA replication errors [77]. Currently, autosomal dominant (AD) is considered as the only disease inheritance mode for PRRT2 traits in OMIM, although preliminary evidence from case reports suggest that PRRT2 can cause a more severe NDD through a biallelic pathogenic mechanism and an AR inheritance model [78]. Our findings in Subject #5 provide further support for the contention of a new rare disease trait type, AR versus AD, and inheritance mechanism due to PRRT2 biallelic variation. Moreover, these observations may also highlight a potential compound inheritance gene dosage (CIGD) model that explains penetrance of certain neurological phenotypes observed in patients with the 16p11.2 deletion; a similar biallelic compound inheritance gene dosage model underlies the penetrance of ~10–12% of all congenital scoliosis worldwide [79].
NAHR deletions contribute to recessive disease burden in population-specific patternsAs suggested earlier, the contribution of a given allele to rare recessive disease trait burden is influenced by the composition of other pathogenic alleles from the same gene. Although the genetics and genomics fields are beginning to appreciate inter-individual variabilities in NAHR rates associated with alternative genomic structural haplotypes [26, 58, 80] as well as polymorphisms from trans acting factors controlling homologous recombination, such as PRDM9 [20], we currently still assume that NAHR mutation rates at a given locus are relatively constant across different populations and genomic ethnic backgrounds. This potentially leaves the remaining alleles, the small variants, as the major driver for any variability in allelic architecture from different population groups.
To investigate the degree of inter-population variability for small variant recessive alleles, we used ethnic information from gnomAD and conducted the modeling described earlier for four population groups, African (AFR), Latino (AMR), East Asian (EAS), and European (EUR) (Additional file 1: Table S5). Population-specific NIRD scores are compared with the general population to generate ΔNIRD (Fig. 3), which can be used to inform the relative odds ratio for NAHR deletions in rare AR disease traits in the specific populations. These analyses provide preliminary computational confirmation for the suspected population variability in NIRD, which implicates that the precision of NIRD can be improved by “tuning the disease model” with population-specific allelic architecture. In light of these surprising findings, we tentatively propose, i.e., we hypothesize, that a prioritization strategy based on prior knowledge of population allele frequency spectra can be applied to enhance discovery in research study design of genomic sequencing among individuals with large recurrent deletions. We cautiously note that some of the population groups analyzed here may not have a sufficient sample size to allow a complete representation of disease alleles of relatively lower frequency, which may result in overestimation of ΔNIRD when the score is positive. Additional population-specific allele frequency data are warranted to improve the accuracy of these analyses.
Fig. 3
留言 (0)