
記住我




To address the biological significance of hypoxia in early brain development, we focused on deleting Hif1α, a master regulator of cellular response to hypoxia [8,9,10,11]. We crossed Hif1α flox/flox mice [16] with Sox1-Cre+/− mice [26] to selectively ablate Hif1α in early neural progenitor cells, thereby affecting all CNS cells. The resulting Hif1α gene carrying an exon 2 deletion encoded a malfunctioning Hif1α protein that lost the ability to induce the expression of genes containing HRE [16]. We confirmed the tissue specificity of Cre expression driven by Sox1 promoter by generating Sox1-Cre+/−; ROSA26/CAG-floxed STOP-tdTomato+/− reporter mice. Cre expression was readily detected by the red fluorescent signal in developing cortical neurons, but not in vasculature at E16.5, indicating neuroepithelial cell-specific expression of Cre driven by Sox1 promoter (Additional file 1: Figure S1). Thus, we investigated the function of Hif1α-dependent hypoxic response in neural progenitor cells and their progeny during brain development. Heterozygotes (Hif1α flox/wt; Sox1-Cre+/−) were viable and fertile, indicating that ablation of a single Hif1α allele or the expression of Cre did not affect CNS development. In contrast, homozygotes (Hif1α flox/flox; Sox1-Cre+/−) conditional mutants died within several hours after birth. Mice homozygous for the floxed Hif1α allele with a Sox1-Cre allele (Hif1α flox/flox; Sox1-Cre+/−) were used to generate conditional knockout mice, which are hereafter referred to as “KO” mice. Mice homozygous for the floxed Hif1α allele without a Sox1-Cre allele (Hif1α flox/flox; Sox1-Cre−/−) were used as controls and are hereafter referred to as “WT” mice.
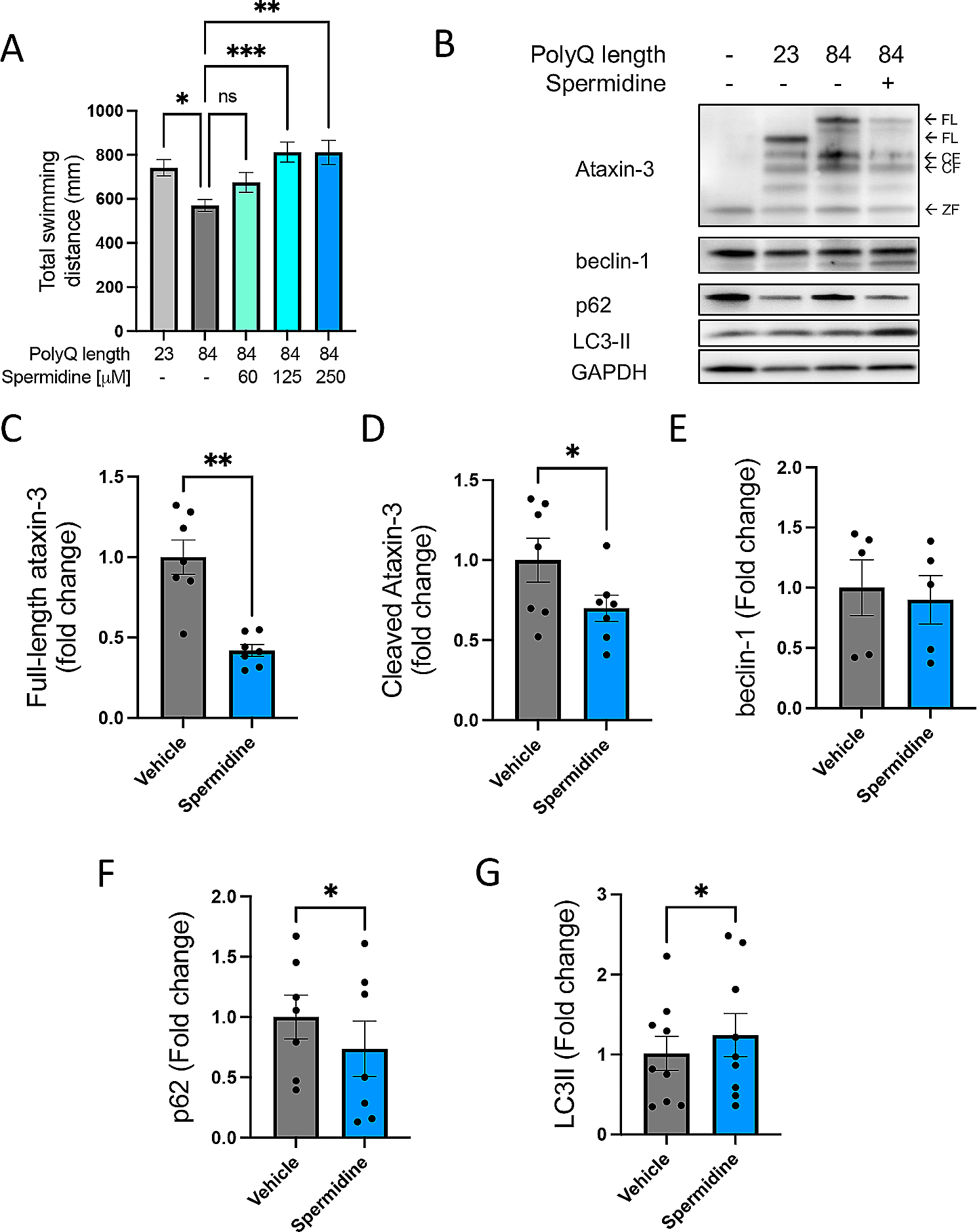
We confirmed the deletion of Hif1α using in situ hybridization with an antisense probe against exon 2. At E8.5, Hif1α mRNA was predominantly expressed in neuroepithelial cells and mesenchymal cells of WT embryos. The expression patterns and levels of Hif1α mRNA in KO embryos were comparable with those in WT embryos (Fig. 1A and B). At E9.5, Hif1α continued to be expressed in neuroepithelial cells and mesenchymal cells in WT embryos (Fig. 1C). In contrast, Hif1α expression diminished in the neural tube of KO embryos, although it remained unchanged in mesenchymal cells at E9.5 (Fig. 1D). To further validate the ablation of Hif1α, the expression of Hif1α and its target genes, Ldha, Aldoa (encoding a glycolytic enzyme) and Epo (encoding erythropoietin), was analyzed using RT-qPCR. Hif1α expression was slightly decreased by loss of Hif1α, while Hif1α target genes were expressed in KO embryos at levels similar to those in WT embryos at E8.5 (Fig. 1E). However, Hif1α expression decreased by approximately 60% in KO embryos relative to that in WT embryos at E9.5 (Fig. 1F), which was consistent with the results of in situ hybridization showing the near-complete loss of Hif1α transcripts in the neuroepithelium (Fig. 1D). Accordingly, the expression of Hif1α target genes was downregulated in E9.5 KO embryos (Fig. 1F; Ldha, reduced by 62%; Aldoa, by 77%; and Epo, by 59%). We isolated total RNA from whole mouse embryos and then prepared cDNA for RT-qPCR. Although it is preferred that total RNA is isolated exclusively from the neuroepithelium, this is technically challenging in early-stage embryos. Therefore, the reduced expression of Hif1α and Hif1α target genes would have been more severe if cDNA prepared from isolated neuroepithelium was used. As controls, we evaluated the expression of EpoR (encoding erythropoietin receptor) and Nqo1 (encoding electron respiratory chain enzyme), which are not dependent on Hif1α. The results showed that their expression was not altered in KO embryos at E9.5 (Fig. 1F); consequently, it was inferred that the decrease in Ldha, Aldoa, and Epo expression levels was not due to a deficit in respiratory metabolism and erythropoiesis, but instead reflected the loss of Hif1α-dependent gene regulation in the early neuroepithelium. Taken together, these results indicate that Hif1α was specifically ablated in neuroepithelial cells as early as E9.5 in KO embryos.
Fig. 1
Conditional ablation of the Hif1α gene. Expression of Hif1α mRNA was detected using whole-mount and cryosection in situ hybridization of E8.5 (A and B) and E9.5 (C and D) embryos. Neural tube is indicated by the dotted line. hm, head mesenchyme; nt, neural tube; Scale bars, 100 μm. Eight (A and B) and four (C and D) independent experiments are performed and one representative image is shown, respectively. Expression levels of Hif1α and its target genes at E8.5 (E) and E9.5 (F) examined using RT-qPCR. The mRNA levels of target genes were normalized to those of Gapdh, and the relative values are presented as bars. White bar, WT; gray bar, KO. Data are the mean ± S.E.M of five embryos. Statistical differences were assessed using Student’s t-test, * p < 0.05. WT, wild type; KO, knockout
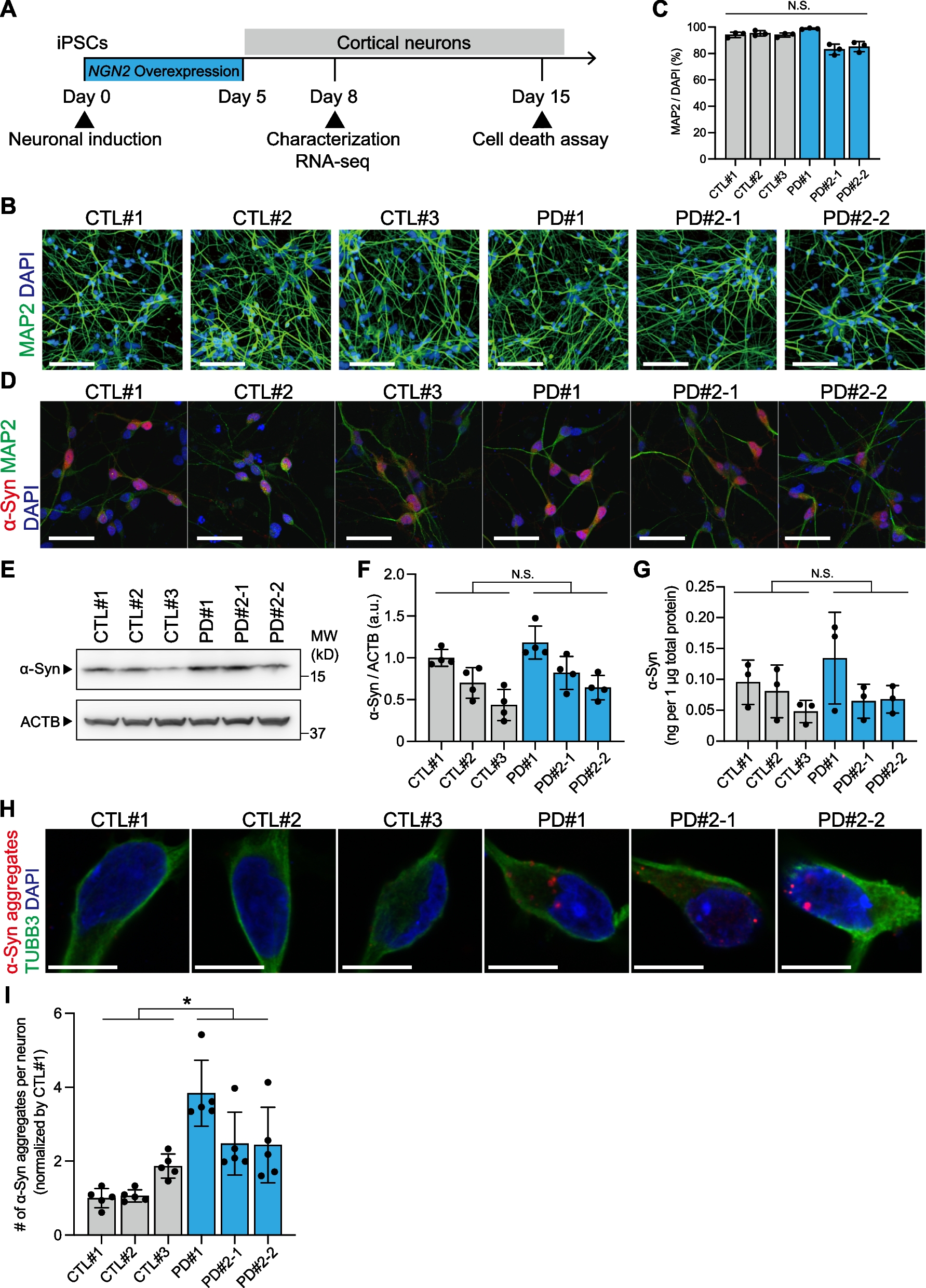
Hif1α ablation causes severe brain defectsNext, we examined the gross morphology of embryos and pups. Although KO embryos were indistinguishable from WT embryos in appearance until E12.5, they exhibited an abnormal-shaped head at E14.5. The length of the head along the sagittal axis was substantially reduced in KO embryos compared to that in WT embryos at E14.5 (Fig. 2A and B, curved two direction arrow). At E16.5, the parietal region of the head was flattened in KO embryos (Fig. 2D, arrowhead), which became more evident by P0 (Fig. 2F, arrowhead). Head abnormalities typically indicate brain hypoplasia; thus, we next examined the morphology of the brain. The brain of KO embryos was smaller than that of their WT counterparts, particularly the telencephalon at E14.5 and E16.5 (Fig. 2G–J). The brain of P0 KO pups exhibited considerable hypoplasia compared to that of WTs. Notably, the cerebrum of KO pups was extremely small compared to that of WT littermates (Fig. 2K and L). Collectively, these results demonstrate the requirement of Hif1α for normal development of the brain, especially that of the cerebrum.
Fig. 2
Conditional ablation of Hif1α in neuroepithelial cells results in a smaller telencephalon. Morphology of the head of WT (A, C, and E) and KO (B, D, and F) mice at E14.5, E16.5, and P0 are shown. Abnormally flattened parietal region is indicated by arrowheads. Scale bars, 1 mm. Morphology of whole-brain of WT (G, I, and K) and KO (H, J, and L) mice at E14.5, E16.5, and P0 are shown. Scale bars, 1 mm. WT, wild type; KO, knockout
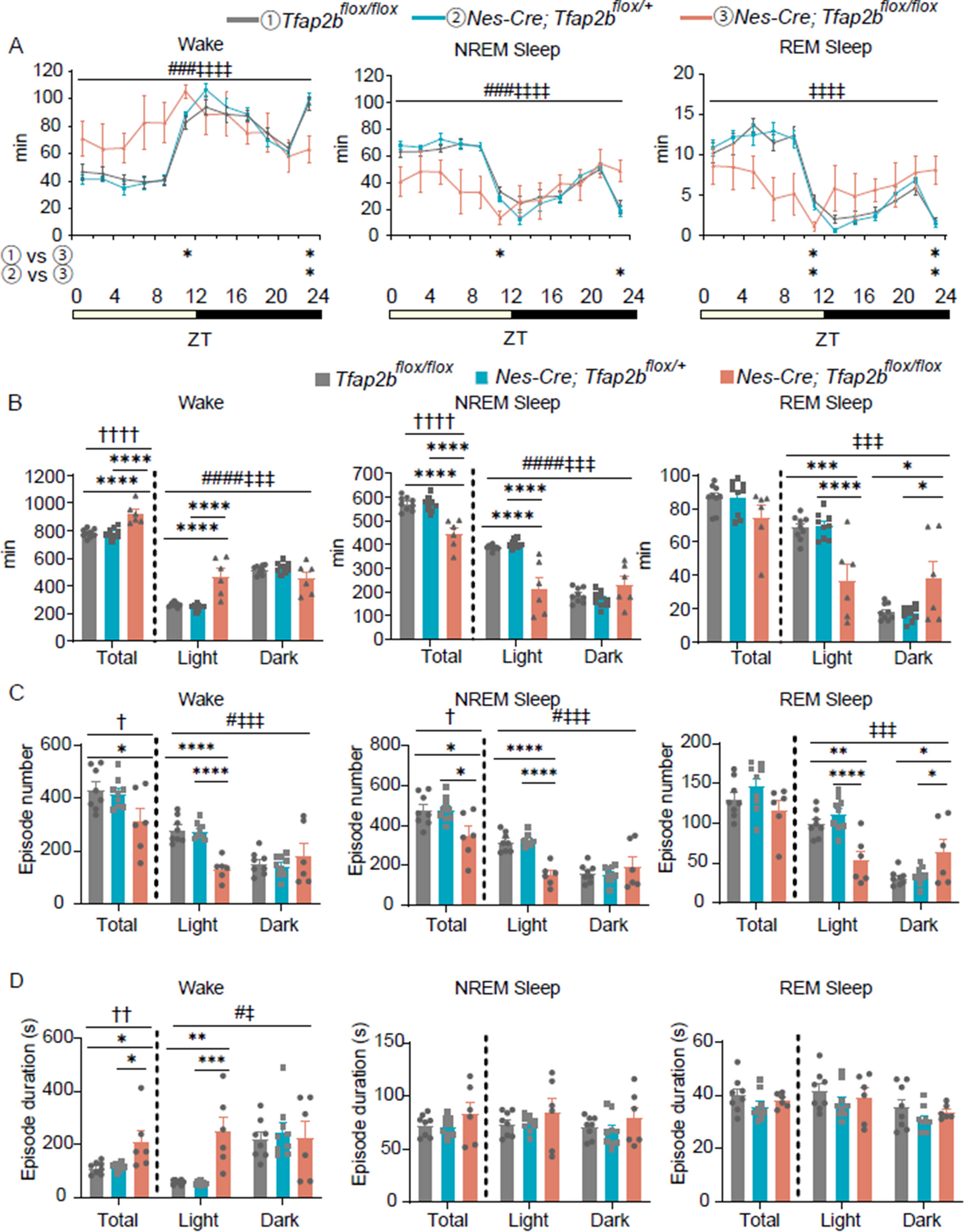
Neurons undergo massive apoptosis in Hif1α-ablated embryosBecause the telencephalon was most acutely affected by the loss of Hif1α, we investigated the role of Hif1α-dependent hypoxia signaling in telencephalic development. The ablation of Hif1α has also been reported to induce apoptosis of neuronal cells in the telencephalon [21, 23]. Thus, we examined apoptosis using immunofluorescent labeling of cleaved caspase3, a marker of apoptotic cells. Some apoptotic cells were detected in the dorsal telencephalon of KO embryos at E12.5 (Fig. 3B). By E14.5, KO embryos displayed enhanced levels of apoptotic cells in the pallium (dorsal telencephalon) and the subpallium (ventral telencephalon) (Fig. 3D). The number of apoptotic cells in KO embryos was further increased in both the pallium and subpallium by E16.5 and E18.5 (Fig. 3F and H).
Fig. 3
Conditional ablation of Hif1α in neuroepithelial cells causes massive apoptosis. Cleaved caspase3+ apoptotic cells (magenta) in coronal sections of the WT (A, C, E, and G) and KO (B, D, F, and H) telencephalon at the indicated embryonic stages are shown. Higher magnification images of the area enclosed by a rectangular dotted line are shown as insets. Scale bars, 200 μm. WT, wild type; KO, knockout. Three (A–D) and four (E–H) independent experiments are performed and one representative image is shown, respectively
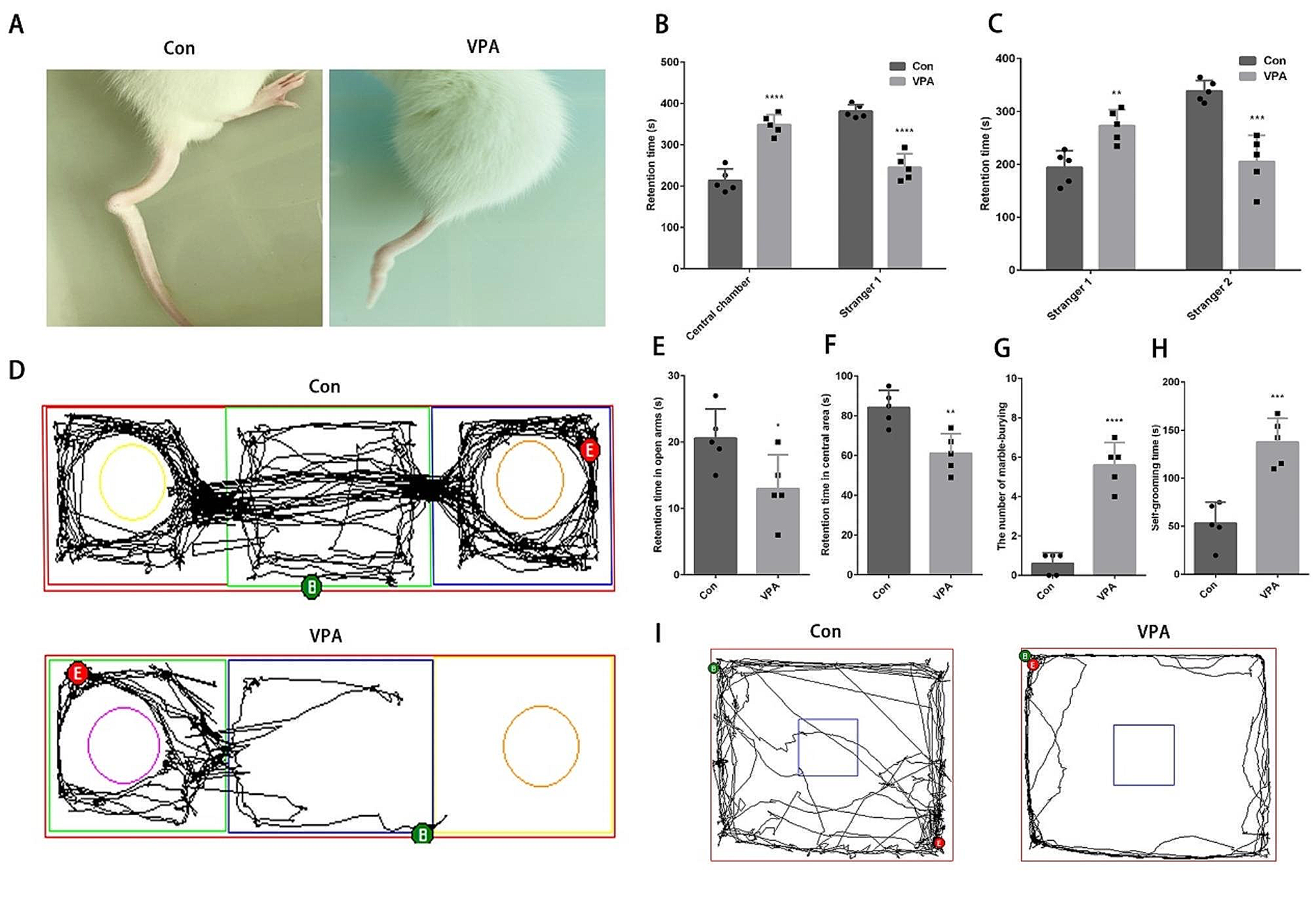
Cerebral cortex layers are disorganized by the loss of Hif1αWe further analyzed apoptosis in the KO cortex at P0. A large proportion of apoptotic cells resided in the Tuj1+ neuronal layer of the cortex, and few apoptotic cells were detected in the intermediate, subventricular, and VZ (Fig. 4B). This observation suggests that Hif1α loss led to the preferential elimination of postmitotic neurons, and consequently led to the reduction of the size of the cerebrum. We counted the number of Satb2+ and Ctip2+ cortical neurons in four arbitrarily defined regions (100 μm-wide) in the P0 cortex. Satb2 is intensely expressed in upper-layer neurons (layers II–IV), whereas Ctip2 is mainly expressed in deep-layer neurons (layers V and VI) in the cortex. The number of Satb2+ and Ctip2+ cells in the P0 cortex was reduced by approximately 88% and 49%, respectively, compared to WT controls (Table 1). This indicated that deep-layer neurons are more sensitive to Hif1α ablation than upper-layer neurons. Remarkably, apoptotic cells formed aggregates in the cortex (Fig. 4B), implying a disruption of the cortical layer formation in the KO cortex. The cortex consists of molecularly distinct Satb2+ upper layer and Ctip2+ deep layer projection neurons in the cerebrum of WT embryos (Fig. 4C). In contrast, each cortical layer was obscured, and Satb2+ and Ctip2+ cells were broadly scattered throughout the KO cortex (Fig. 4D).
Fig. 4
Conditional ablation of Hif1α in neuroepithelial cells causes neuronal apoptosis and consequently disturbs the formation of cortical layers in the cortex. Apoptotic cells and cortical neurons in coronal sections of WT and KO cortices at P0 are detected with anti-cleaved caspase3 and Tuj1 (anti-β-tubulin class III) (A, B), or anti-Satb2 and anti-Ctip2 (C, D) antibodies. Reelin expression in coronal sections of WT and KO cortices at E18.5 is detected with anti-Reelin antibody (E, F). Scale bars, 100 μm (A–F). WT, wild type; KO, knockout. Four (A–D) and three (E and F) independent experiments are performed and one representative image is shown, respectively
Table 1 The number of Satb2- and Ctip2-positive cells in P0 cortexThe disorganized cortical layers was reminiscent of the phenotype of Reelin-deficient mice, reeler [27]. Cajal-Retzius (C-R) cells, which occur in the most superficial marginal zone layer of the cerebral cortex, regulate cortical layer formation through the secretion of Reelin [28, 29]. Therefore, the Sox1-Cre-mediated deletion of Hif1α during early telencephalic development may have affected the survival of C-R cells. However, C-R cells were detected in the most superficial layer (layer I) of the cortex, underneath the pia, in both WT and KO telencephalons (Fig. 4E and F), suggesting that the Reelin-mediated guidance of postmitotic neurons is intact in the KO cortex.
Neuronal migration is impaired in Hif1α-ablated cortexTo explore the mechanism inducing neuronal apoptosis by loss of Hif1α, we examined whether upper- or deep-layer neurons undergo apoptosis via immunofluorescent staining using anti-cleaved caspase3 and anti-Satb2 or anti-Ctip2 antibodies (Fig. 5A–D). We observed that many Ctip2+ cells were co-labeled with cleaved caspase3 (Fig. 5D, inset). The number of Satb2+ /cleaved caspase3+ double-labelled cells was lower than that of Ctip2+ /cleaved caspase3+ cells in the E18.5 KO cortex (Fig. 5B, inset). Specifically, approximately 8.4% and 35.1% of total cleaved caspase3+ cells were co-labeled with Satb2 and Ctip2, respectively (Fig. 5E). This demonstrated that deep-layer neurons preferentially undergo apoptosis in the absence of Hif1α, which was consistent with data showing a predominant reduction of deep-layer neurons in the KO cortex (Table 1). Approximately 56% of all cells were cleaved caspase3+ cells, thereby explaining the severe loss of layer marker expression.
Fig. 5
Predominant apoptotic elimination of Ctip2+ deep-layer neurons affects cortical layer formation. Apoptotic cells and Satb2+ (A, B) or Ctip2+ (C, D) cells in coronal sections of the KO cortex at E18.5 are shown. Higher magnification images of the area enclosed by rectangular dotted line are shown as insets (B, D). Satb2+ cells accumulating at the apical side of the apoptotic cell aggregate are indicated by yellow arrowheads. Ctip2+ cells distributed near the pial surface are indicated by white arrowheads. Scale bars, 100 μm (A–D). Three independent experiments are performed and one representative image is shown, respectively. The percentage of cleaved caspase3 + and Satb2+ or Ctip2+ cells is presented as a bar graph (E). Data are the mean ± S.E.M of six sections. Statistical differences were assessed using Student’s t-test, * p < 0.05
Many Satb2+ upper-layer neurons were detected around apoptotic cell aggregates, especially on the apical side of the KO cortex (Fig. 5A, yellow arrowheads). Consistent with this, Satb2+ cells were sparsely distributed on the pial surface side of apoptotic cell aggregates. Conversely, Ctip2+ neurons were abnormally detected near the pial surface side where the upper layers were formed in the normal cortex (Fig. 5C, white arrowheads). The cerebral cortex consists of six neuronal layers that develop in an inside-out manner, i.e., early-born neurons settle in the deep layers, whereas late-born neurons migrate through the deep-layer neurons and form more superficial layers (upper-layers) [30]. Thus, we hypothesized that the migration of the upper-layer neurons from the VZ to the pial surface was impeded by apoptosis of deep-layer neurons, resulting in a partial reversal of the cortical layer in the KO cortex. To test this hypothesis, we performed a BrdU birthdating assay. To label the upper-layer neurons, BrdU was injected into pregnant mice at E14.5, when upper-layer neurons are being born, and then the position of BrdU-labeled neurons were compared between KO and WT cortices at E18.5. A large proportion of BrdU+ neurons were located in the upper-layers of the WT cortex (corresponding to bins 1 and 2; Fig. 6A and B). In contrast, many BrdU+ neurons were detected in the deeper-layers of the KO cortex. Importantly, most BrdU+ neurons were negative for apoptosis markers (Fig. 6C and D), which is consistent with the results shown in Fig. 5B and E. The distribution of BrdU+ neurons born at E14.5 was quantified in five arbitrarily defined regions (500 μm-wide) in the cortex. In the WT cortex, approximately 72% of BrdU+ neurons were present in the upper-layers (bins 1 and 2). In contrast, the percentage of BrdU+ neurons in the presumptive area of the upper-layers (bins 1 and 2) was reduced to approximately 36% in the KO cortex (Fig. 6G). Approximately 42% of BrdU+ neurons populated the middle area (bins 3 and 4) in the KO cortex, where a large number of apoptotic cells were observed. In contrast, approximately 21% of BrdU+ neurons were detected in the same region of the WT cortex (Fig. 6G). Notably, the total number of BrdU+ neurons in the KO cortex was comparable with that of BrdU+ neurons in the WT cortex, suggesting that the proliferation of neural progenitors of upper-layer neurons was not altered by the loss of Hif1α (Fig. 6H). Taken together, these results strongly support our hypothesis that the migration of upper-layer neurons was impeded by massive apoptosis of deep-layer neurons in the KO cortex.
Fig. 6
Neural migration of upper-layer neurons is impaired by loss of Hif1α. Apoptotic cells and BrdU-incorporated neurons in coronal sections of WT and KO cortices at E18.5 are detected with anti-cleaved caspase3 and anti-BrdU antibodies (A–D). Scale bars, 100 μm (A–D). Three independent experiments are performed and one representative image is shown, respectively. The percentage of BrdU+ cells in the cortex (bins 1–5, 500 μm-wide) is presented as a histogram (G). The total number of BrdU+ cells in the cortex (500 μm-wide region) is presented as a bar graph (H). Gray bar, WT; black bar, KO. Data are the mean ± S.E.M of four sections. Statistical differences were assessed using Student’s t-test, * p < 0.05. WT, wild type; KO, knockout
Postmitotic neurons migrate towards the pia along the radial glial cell fiber spanning the length of the developing cortical plate, and is important for proper ‘inside-out’ neural migration [27, 28, 30]. Almost radial glial fibers spanned the entire cortex in both of WT and KO telencephalon (Additional file 2: Figure S2). This result suggests that the radial glial fibers-dependent neural migration is intact in the KO cortex.
Possible mechanism underlying the role of Hif1α in cortical developmentPrevious studies demonstrated that vascular endothelial growth factor (encoded by Vegf), a downstream target of Hif1α, functions not only as an angiogenic growth factor but also as a neurotrophic factor [31, 32]. It has also been demonstrated that neuronal cell-specific Vegf homozygous knockout mice die within 24 h after birth due to the dysmorphogenesis of the cortex caused by massive neuronal apoptosis [33, 34]. Moreover, as apoptotic cells seem to accumulate in the middle area of the cortex [33], the Vegf homozygous knockout phenotype resembles that of our Hif1α homozygous knockout mice. We therefore investigated if the reduced Vegf expression was associated with reduced survival of deep-layer neurons upon Hif1α loss. Vegf is expressed in the ventral layer of the brain at E14.5 [35]. First, we analyzed Vegf expression levels in WT and KO telencephalons at E13.5, when deep-layer neurons are born. To analyze this, the dorsal part of the telencephalon, including the cortex, was surgically isolated, and RNA was prepared for RT-qPCR. We confirmed a significant reduction in Vegf expression in the cortex of KO mice (Fig. 7A). Next, we examined whether Vegf signaling activity is required for the survival of deep-layer neurons. Although the suppression of Vegf receptor function is the ideal method to inhibit the input of Vegf signaling, known receptors for Vegf, such as Flk1, Flt1, Nrp1, and Nrp2, do not appear to be involved in cortical development and thus unlikely targets to manipulate Vegf signaling in the developing brain [34, 36,37,38]. Therefore, we took advantage of the ligand-binding domain of the Vegf receptor, sFlt1, to interfere with Vegf signaling [39]. It has been reported that the overexpression of sFlt1 inhibits Vegf signaling in vivo [40]. We thus hypothesized that secreted sFlt1 functions as a decoy Vegf receptor and inhibits paracrine and/or autocrine Vegf signaling inputs to neural progenitors and/or neurons of deep layers. The expression construct of mouse sFlt1 was introduced into neural progenitor cells of E13.5 WT embryos using in utero electroporation along with the EGFP expression construct. At E18.5, numerous EGFP + sFlt1-overexpressing cells were co-labeled with apoptotic markers, while no apoptosis was observed in the control cortex (Fig. 7B–F). However, we could not detect apoptotic cell aggregates, which may be due to the variation in transfection efficiency of sFlt1 (Fig. 7D and E). Next, we investigated cortical layer formation in the sFlt1-transfected cortex. Satb2+ neurons were dispersed in the apical side of the cortex where deep-layer neurons normally settle in the control cortex (Fig. 7G, H, K, L). The Ctip2+ neuron layer was considerably shifted to the pial surface in the transfected region of sFlt1, consistent with the accumulation of upper-layer neurons in the deep layers of the KO cortex (Fig. 7I, J, M, N). In contrast, apoptosis and cortical layer defects were not induced when the sFlt1-expressing construct was electroporated into neural progenitor cells at E15.5 (Fig. 7O–T), suggesting that Vegf signaling is not required for the survival of upper-layer neurons. Taken together, inhibition of Vegf signaling by the overexpression of sFlt1 mimics cortical development defects seen in KO embryos.
Fig. 7
Vegf signaling is required for the survival of deep-layer neurons and proper cortex formation. Vegf expression level was quantified using RT-qPCR (A). The mRNA levels of Vegf mRNA normalized to that of Gapdh. The relative values are presented as a bar graph. White bar, WT; gray bar, KO. Data are the mean ± S.E.M of 3 embryos. Statistical differences were assessed using Student’s t-test, * p < 0.05. pCI (control) or sFlt1-pCI (sFlt1) plasmid was electroporated into neural progenitor cells at E13.5 (B–E, G–N). Apoptotic cells (magenta) and transfected cells (green) are detected with anti-cleaved caspase3 and anti-EGFP antibodies, respectively, in coronal sections of the cortex at E18.5 (B–E). The total number of cleaved caspase3+ cells in the cortex (500 μm-wide region) is presented as a bar graph (F). White bar, control; gray bar, sFlt1. Data are the mean ± S.E.M of five sections. Statistical differences were assessed using Student’s t-test, * p < 0.05. Indicated cortical neurons (magenta) and transfected cells (green) are detected with anti-Satb2 (G, H, K, L) or anti-Ctip2 (I, J, M, N) and anti-EGFP antibodies, respectively, in coronal sections of the cortex at E18.5. Abnormal localization of cortical neurons is indicated with the asterisk. sFlt1-pCI plasmid was electroporated into neural progenitor cells at E15.5 (O–T). Apoptotic cells (magenta) and transfected cells (green) with anti-cleaved caspase3 and anti-EGFP antibodies, respectively, in coronal sections of the cortex at E18.5 (O and P). Indicated cortical neurons (magenta) and transfected cells (green) with anti-Satb2 (Q and R) or anti-Ctip2 (S and T) and anti-EGFP antibodies, respectively, in coronal sections of the cortex at E18.5. Scale bars, 100 μm (B–E, G–T). Three (B–E, O–T) and four (F–M) independent experiments are performed and one representative image is shown, respectively
As we postulated that neuronal apoptosis is due to the decrease in Vegf levels in the KO cortex, it is important to identify the source of Vegf in the telencephalon. Unfortunately, we could not detect the expression of Vegf mRNA and Vegf protein in the telencephalon using in situ hybridization and immunofluorescence (data not shown). Instead of that, we tried to determine whether Vegf functions in cell-autonomous or non-cell-autonomous manner. The expression construct of Cre recombinase was introduced into E13.5 Hif1α flox/flox telencephalon using in utero electroporation. At E18.5, EGFP + Hif1α-ablated cells did not undergo apoptosis (Fig. 8A), suggesting the non-cell-autonomous function of Vegf for the survival of deep-layer neurons. Notable, many EGFP + Hif1α-ablated cells were localized more apical side compared to EGFP+ cells in control cortex (Fig. 8B and E). This result indicates that, besides regulating the survival of deep-layer neurons in non-cell-autonomous manner, Hif1α functions cell-autonomously in neuronal migration.
Fig. 8
Vegf functions as the survival factor for deep-layer neurons in non-cell-autonomous manner. pEGFP-CAGS (control) or pCAG-Cre:GFP (Cre:GFP) plasmid was electroporated into neural progenitor cells in one side of telencephalic semisphere of Hif1α flox/flox embryo at E13.5. Cleaved caspase3+ apoptotic cells (magenta) and EGFP+ transfected cells (green) were detected in coronal sections of the cortex at E18.5. Scale bars; 100 μm. Three independent experiments are performed and one representative image is shown, respectively
留言 (0)