Chemicals used in experiments and ethical approval
All chemicals and media were purchased from ThermoFisher (Waltham, MA, USA) or Sigma-Aldrich Chemical Company (St. Louis, MO, USA), unless otherwise specified. Collection of synovial fluid (SF) specimens and PBMCs was performed after obtaining informed consent from patients and volunteers (Approval number GNUH 2012-05-009). The protocol for animal experiments in CIA mice was approved by the Animal Center for Biomedical Experimentation at Gyeongsang National University (GNU-131209-M068).
Collection of SF and establishment of SF-MSCs from RA patients
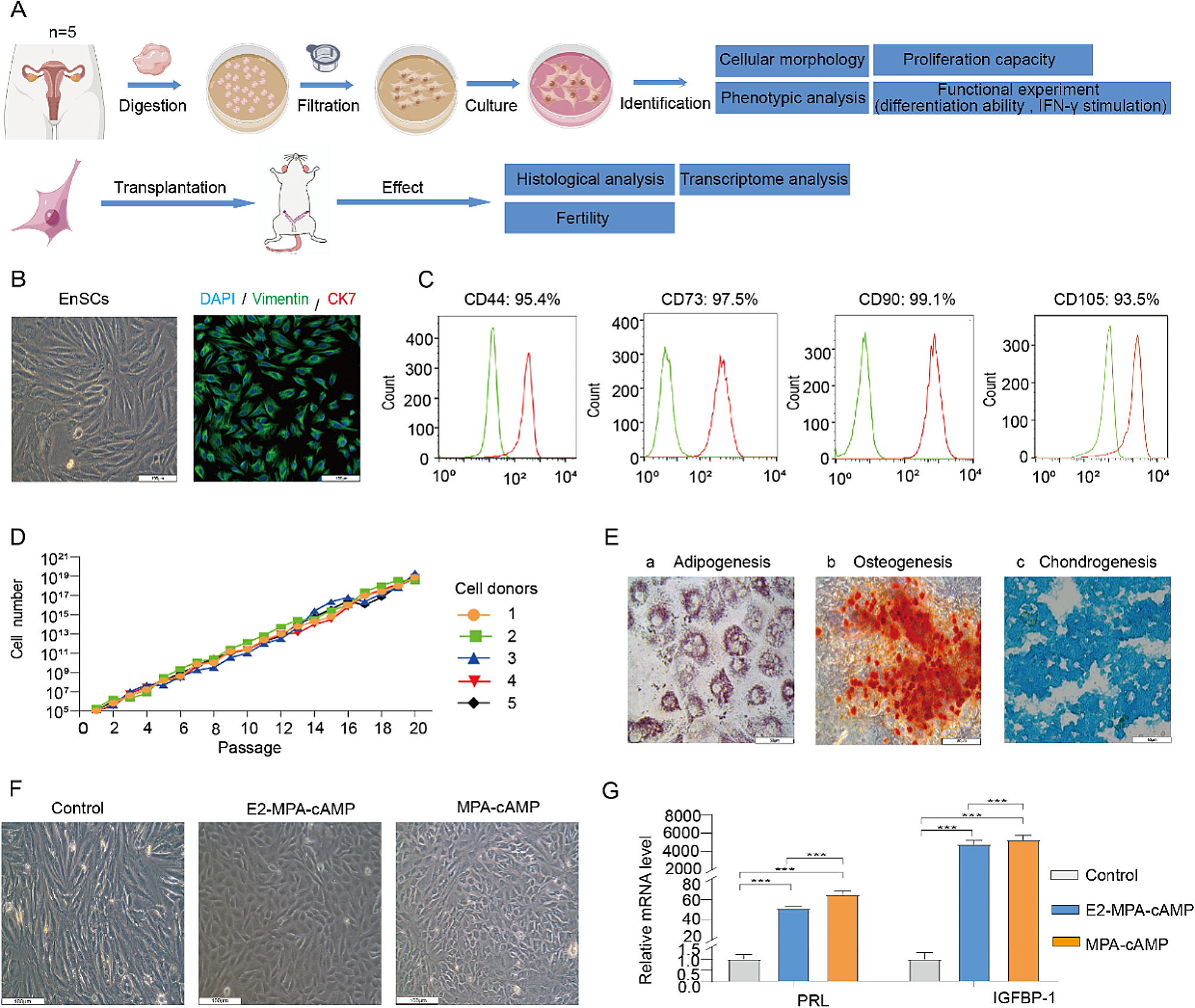
Control SF was obtained from donors without evidence of inflammatory joint disease. SF from the RA groups was obtained from the joints of RA patients who were divided into early (E-RA, disease duration < 2 years) or long-standing (L-RA, disease duration > 10 years) groups. Then, the SF-MSCs were divided into three groups: CTL-SF-MSCs (n = 10), E-SF-MSCs (n = 9) and L-SF-MSCs (n = 12). The clinical histories of the RA patients are presented in Table 1. Cells were isolated from the aspirated and cultured SF and were processed as previously described [3]. The SF specimens were filtered through a 40-μm nylon cell strainer (BD Falcon, NJ, USA) to remove debris and centrifuged at 400×g for 10 min. The supernatants were stored at −80°C until the inflammatory cytokine analysis, while the cell pellet was resuspended and explanted onto 35 mm dishes (Nunc, Roskilde, Denmark). The cells were allowed to adhere for 2 days in culture medium before non-adherent cells were discarded. The adherent cells were cultured with advanced Dulbecco’s modified Eagle’s medium (ADMEM) supplemented with 10% fetal bovine serum (FBS), 1% GlutaMax, 10 ng/mL bFGF, and 1% penicillin and streptomycin (10,000 IU and 10,000 μg/mL) at 36.5°C in a humidified incubator with 5% CO2. The expanded cells were passaged four times before use for further analysis.
Table 1 Demographic and disease characteristics of donorsCharacterization of SF-MSCs
Expression of MSC-specific cell surface molecules in SF-MSCs were validated in triplicate by flow cytometry using a BD FACS Calibur instrument (BD Biosciences, NJ, USA). A total of 1 × 104 cells were harvested and fixed with 4% paraformaldehyde at 4°C. All antibodies were diluted (1:200) with 1% bovine serum albumin (Table S1). Fluorescein isothiocyanate (FITC)-conjugated primary antibodies were incubated with the harvested cells for 1 h, with mouse IgG1-FITC used as an isotype control. Approximately ~80% of confluent SF-MSCs differentiated into adipocytes and osteoblasts after 3 weeks. Adipogenesis was induced with Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 100 mM indomethacin, 10 mM insulin, and 1 mM dexamethasone. Then, adipogenesis was confirmed by intracellular lipid vacuole staining with 0.5% Oil red O solution and by gene expression (FABP4 and PPARγ). Osteogenesis was induced with DMEM supplemented with 10% FBS, 200 mM ascorbic acid, 10 mM β-glycerophosphate, and 0.1 mM dexamethasone. Then, osteogenesis was determined by the accumulation of calcium deposits visualized with Alizarin-red S solution and by gene expression (ON and OCN). For chondrogenesis, 1 x 106 SF-MSCs were cultured for 3 weeks in 15 mL tubes containing STEMPRO Osteocyte/Chondrocyte basal medium supplemented with 10% chondrogenesis supplement. Cell pellets were embedded in paraffin, cut into 5 mm sections, and stained with 1% Alcian blue and 0.1% nuclear fast red counterstain to confirm proteoglycan synthesis. Chondrogenesis was also verified by gene expression analysis (COL2 and COL10A1). The protocol for the gene expression analysis is described below.
Gene expression by quantitative PCR (qPCR)
qPCR was used for gene expression studies to determine pluripotency (Oct3/4, Sox2, and Nanog), apoptosis (Bax, Bak, p53, Bcl2, and Birc), differentiation (FABP4, PPARγ, ON, OCN, COL2, and COL10A1), and the expression of hypoxia-related genes (GLUT1, LDHA, LOX, and PGK1). Three replicates of each sample were analyzed by qPCR. Relevant primer information is displayed in Table S2. The total RNA was extracted using an RNeasy Minikit (Qiagen, CA, USA) and quantified using an OPTIZEN 3220 UV BIO spectrophotometer (Mecasys, Sungnam, Korea). Next, cDNA synthesis was performed from 1 μg total RNA using an Omniscript Reverse Transcription Kit (Qiagen) with a oligo dT primers at 60°C for 1 h. qRT-PCR was performed using a Rotor Gene Q qRT-PCR instrument (Qiagen) with Rotor-Gene 2× SYBR Green mix (Qiagen), 2 μL cDNA per reaction, and 0.5 mM forward and reverse primers. The qPCR program settings included of pre-denaturation (95°C for 10 min), 45 PCR cycles (95°C for 10 s, 60°C for 6s, and 72°C for 4 s), melting curve analysis (60°C to 95°C ramp, 1°C per seceond ramp rate) and cooling (40°C for 30 s). Transcript levels of all target genes were normalized against those of TBP, which is a stable reference gene in human MSCs [12].
Proliferation and cell cycling in SF-MSCs
Vybrant MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] Cell Proliferation Assays (Molecular Probes, Eugene, OR, USA) were used to evaluate SF-MSC proliferation. Absorbance at 540 nm was measured using a microplate reader (Molecular Devices). To analyze cell cycle changes, SF-MSCs were fixed with 70% ethanol, stained with 10 μg/ml propidium iodide (PI) solution, and evaluated using flow cytometry.
Senescence-associated β-galactosidase activity staining
Cellular senescence was evaluated using Senescence β-Galactosidase Staining Kits (Cell Signaling Technology, Danvers, MA, USA). SF-MSCs were fixed for 15 min in fixation solution at room temperature, stained with β-Galactosidase staining solution, and incubated at 37°C overnight. To measure β-galactosidase activity, Mammalian β-Galactosidase Assay Kits (ThermoFisher, Rockford, IL, USA) were used. SF-MSCs were harvested, incubated with M-PER reagent for 10 min, and centrifuged for 10 min at 27,000xg. The supernatant was transferred into 96-well plates and treated with β-galactosidase reagent for 30 min at 37°C. The optical density was determined at 405 nm using a microplate reader (Molecular Devices).
Evaluation of telomere length and telomerase activity
Telomere lengths in SF-MSCs were investigated using a nonradioactive chemiluminescent TeloTAGGG telomere restriction fragment (TRF) length assay kit (Roche, Indianapolis, IN, USA).
Suppression of PBMC proliferation by SF-MSCs
Human PBMCs were isolated from healthy donors (n = 6) by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare, Uppsala, Sweden). PBMCs were resuspended in RPMI 1640 complete medium supplemented with 10% FBS, 1% penicillin, and 1% streptomycin (10,000 IU and 10,000 μg/mL). Then, the cultures were stimulated with 1 μg/mL PHAL to activate T-cell proliferation. The PHAL-activated PBMCs (6.25 × 103 cells/well) were seeded in a 96-well plates. After 12 h, 10 μg/mL Mitomycin-C (Sigma-Aldrich, USA) was added 2 h to inhibit cell proliferation. The PHAL-activated PBMCs were co-cultured for 5 days in a 96-well plate with pre-seeded SF-MSCs at PBMC to MSC ratios of 1:4, 1:2, and 1:1 before the addition of 5-bromo-2-deoxyuridine (BrdU). PBMC proliferation was evaluated using a Cell Proliferation ELISA, BrdU (colorimetric) Kit (Roche Diagnostics, Mannheim, Germany).
Analysis of cytokine levels in SFs and SF-MSCs
The frozen SF supernatant samples were thawed and used to evaluate inflammatory cytokine levels. The levels of tumor necrosis factor (TNF)-α and interleukin (IL)-1β in the SF were determined with Quantikine ELISA kits (R&D Systems, Minneapolis, MN, USA). Briefly, standards and samples were incubated in wells pre-coated with the respective human primary antibody. The resulting antigen-antibody complexes were detected using human TNF-α or IL-1β conjugated to horseradish peroxidase, and the conjugate was quantified by a colorimetric reaction with 3,3′,5,5′-tetramethylbenzidine substrate. The resultant color intensity was read at 450 nm using a microplate reader (Molecular Devices). For SF-MSCs, 6.25 × 103 cells/well were cultured in 96 well plates in serum-starvation medium (1% FBS in ADMEM), followed by supplementation with human recombinant TNF-α (50 ng/mL; R&D Systems) for 2 days to activate inflammatory cytokine production. After collecting the supernatant, the levels of matrix metalloproteinases (MMPs; MMP-1, MMP-3, and MMP-13) and other cytokines [IL-6 and indoleamine-pyrrole 2,3-dioxygenase (IDO)] were analyzed in the same manner as SF samples. All samples were assayed in duplicate and the concentration of target proteins in each sample was determined by interpolation from a standard curve.
SF-MSC administration to CIA mice
Injection of MSCs into CIA mice was conducted as previously described [3]. Briefly, pathogen-free male DBA/1 mice (7–9 weeks old; Orient Bio, Seoul, Korea) were immunized with 100 μg bovine type II collagen (Chondrex, Redmond, WA, USA) emulsified in complete Freund’s adjuvant (CFA, Chondrex) by injection into the intradermal region of the tail on day 0. Mice received a booster immunization of an equal volume of bovine type II collagen and incomplete Freund’s adjuvant (IFA, Chondrex) on day 21. The experiment included 4 groups (n = 8 per group): a PBS injection control, and CTL-SF-MSC-, E-SF-MSC-, or L-SF-MSC-injected groups. SF-MSCs were intraperitoneally injected on day 21 and for five consecutive days with 200 μL PBS or SF-MSCs (5 × 106 cells in 200 μL PBS). Clinical arthritis scores (0–4 scale) were evaluated for each limb in accordance with a well-defined standard. The total possible score was 16. To measure hind paw thickness, a caliper was placed across the ankle joint at the widest point. On day 48, CIA mice were sacrificed by cervical dislocation. The hind paws were scanned with a SkyScan 1076 micro-CT apparatus (Bruker, Kontich, Belgium) and reconstructed into a three-dimensional structure with a voxel size of 18 μm using NRecon and CT Analyzer software (Bruker). Joint tissue specimens from CIA mice were fixed with 10% formalin, decalcified for 3–4 weeks in 10% EDTA, and embedded in a paraffin block. Joint sections (5 μm) were stained with hematoxylin and eosin (H&E), Safranin O, or tartrate-resistant acid phosphatase (TRAP) to evaluate articular inflammation, cartilage damage, and TRAP-positive multinucleated cells (osteoclasts), respectively. The total number of TRAP-positive multinucleated cells containing three or more nuclei was counted in 10 areas of each CIA mouse ankle [3, 13].
Induction of the RA-like inflammatory milieu
Because both low partial oxygen pressure (hypoxia) and inflammation are relevant features in the synovial joints of RA patients [14], an in vitro RA-like inflammation milieu was induced in E-SF-MSCs to explore whether immunomodulatory properties and senescence were altered in inflammation-exposed SF-MSCs. E-SF-MSCs were cultured in normal culture conditions for 3 days with in various gas compositions: 21% O2, 5% CO2, and 74% N2; or 3% O2, 5% CO2, and 92% N2 in a 95% humidified atmosphere. The cells were maintained in multi-gas incubators (ASTEC, Fukuoka, Japan) to reflect normoxia or hypoxia. In addition, the media for hypoxic E-SF-MSCs was supplemented with 20 ng/mL TNF-α and 20 ng/mL IL-1β (R&D Systems) as representative inflammatory cytokines. Both E-SF-MSCs and L-SF-MSCs cultured in normoxic conditions were used as controls.
Western blot analysis
The induction of hypoxia was validated by upregulated hypoxia-inducible factor 1-α (HIF1α) expression. Cell extracts of E-SF-MSCs with or without hypoxia were prepared with RIPA buffer supplemented with Halt Protease Inhibitor Cocktail (Pierce Biotechnology, Rockford, IL, USA). The total protein concentration in the cell extracts was quantified using Bicinchoninic Acid Protein Assay Reagent Kits (Pierce Biotechnology). A 25-μg aliquot from each sample was fractionated by 10% SDS-PAGE and was transferred onto a polyvinylidene difluoride membrane (Millipore, Darmstadt, Germany). The membranes were blocked with 0.1% bovine serum albumin (BSA), incubated with anti-HIF-1α or anti-GAPDH primary antibodies (1:100 dilution with BSA) at 4°C overnight, incubated with horseradish peroxidase-conjugated secondary antibodies (1:3,000 dilution with BSA) at RT for 1 h, and detected using a chemiluminescence assay (Amersham Biosciences Corp, Piscataway, NJ, USA) with X-ray film for visualization.
Apoptosis assays
The proportion of apoptosis in E-SF-MSCs with or without the induction of an RA-like inflammatory milieu was determined using an Annexin V-FITC Apoptosis Detection Kit (Invitrogen, Eugene, OR, USA). For these assays, 1 × 104 cells were harvested, washed twice in PBS, and resuspended in 200 μL binding buffer. Then, the cells were treated with 10 μL Annexin V stock solution, incubated at 4°C for 30 min, counterstained with PI, and analyzed by flow cytometry (BD FACS Calibur).
IDO activity measurements
IDO activity was measured as previously described [15]. Cultures were harvested and 2.5 × 104 E-SF-MSCs with or without induction of an RA-like inflammatory milieu were cultured for 4 days. Then, the cells were supplemented with 100 μM L-tryptophan (Sigma) for 4 h. The supernatant was harvested and mixed with 30% trichloroacetic acid (Sigma) before an additional incubation at 50°C for 30 min. This solution was diluted in Ehrlich reagent (1:1) (Sigma, USA), and the optical density was measured at 492 nm using a microplate reader (Molecular Devices). Serially-diluted l-kynurenine (Sigma) made with fresh culture medium was used as the standard.
Statistical analysis
Statistical significance was analyzed using paired T tests, one-way analysis of variance (ANOVA), and Tukey’s multiple comparison tests followed by Games-Howell post hoc analysis using SPSS 21.0 (IBM, Armonk, NY. USA). All data are presented as mean ± standard deviation (SD). P < 0.05 was considered significantly different.






留言 (0)