
記住我
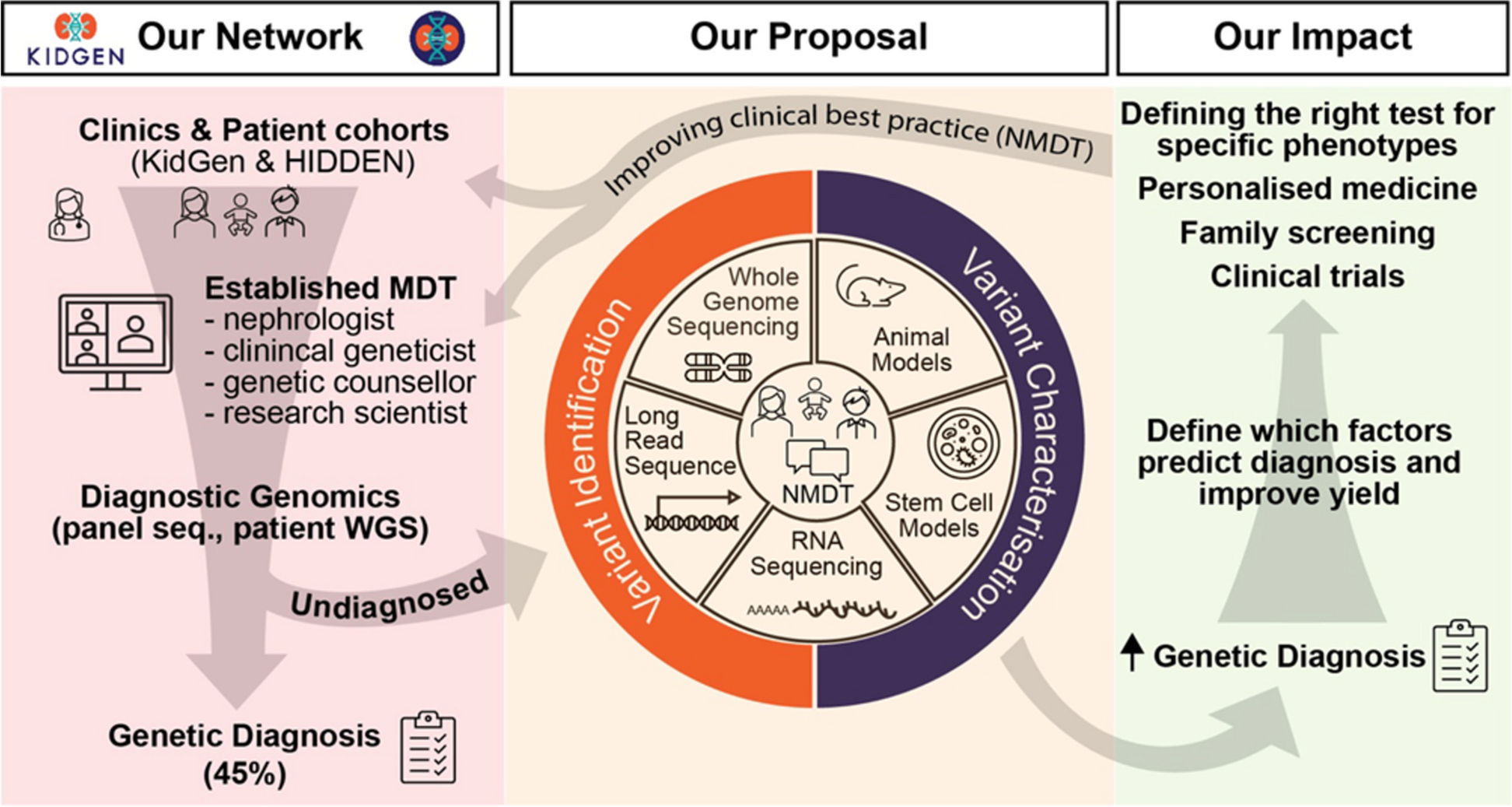



The KidGen National Kidney Genetic Study will operate under a hierarchical structure with five purpose-specific entities meeting at different frequencies. The Governance Committee (9 members) convenes semi-annually and holds the highest authority, overseeing critical decisions. The Steering Committee (23 members) meets every two months to translate the Governance Committee's direction into actionable plans. The National Multidisciplinary Team (nMDT; variable number with a quorum of 5) conducts monthly meetings to discuss proposed cases and enhance knowledge exchange, collaboration, and recruitment of patients nominated by KGCs. There are two working groups: the Clinical Working Group (CWG; 7 members) and the Functional Genomics Group (FGG; 9 members). These two groups focus on specific clinical and discovery areas and report to the Steering Committee. The CWG meets weekly to review all cases nominated for the study against the study inclusion/exclusion criteria, engage with clinics, discuss individual patients, and review the project's operational progress. Additionally, the CWG holds bi-monthly meetings with the FGG to discuss preliminary results from variant analysis and consider options for functional modelling if required. This multilevel structure ensures strategic direction and efficient project execution by maintaining effective collaborative communication.
Study overviewWe aim to recruit approximately 200 families with suspected unexplained GKD after standard testing from our KGCs and existing KidGen research cohorts. These families will undergo comprehensive genomic analysis facilitated by a team of specialists (nMDT). Our approach utilises phenotype-driven research genomics to identify the genetic basis of GKD in each family. Research genomic sequencing will be undertaken, with short-read whole genome sequencing used as the baseline testing for most probands, including family members, where relevant. Bioinformatic processing will be conducted through the Centre for Population Genomics and phenotype-driven variant analysis via an Australian instance of the seqr platform [15], a collaborative platform designed explicitly for analysing rare disease genomic data.
Furthermore, the existing whole-genome sequencing (WGS) and whole-exome sequencing (WES) data from undiagnosed families previously recruited to KidGen studies [9] will be re-analysed using novel analytical tools developed by the Centre for Population Genomics (CPG) at the Garvan Institute of Medical Research/Murdoch Children's Research Institute (MCRI) and other partners. We will also employ advanced modelling techniques for the identified variants of uncertain significance (VUS), including animal and cellular GKD models, to validate their role in disease development. Functional modelling will also be applied to assess novel gene-disease associations. Our comprehensive strategy, detailed in Fig. 1, intends to maximise the chances of a conclusive genetic diagnosis. Crucially, the study will collect data on the resourcing required to achieve diagnosis to address the translation of such a pipeline to standard clinical diagnostics.
Fig. 1
KidGen national kidney genomics program: improving diagnostic outcomes for Australian families with genetic kidney disease. The KidGen network is a nationwide initiative led by the national multidisciplinary team (nMDT), coordinating kidney genetic clinics (KGCs) across Australia. This collaborative framework offers subsidised clinical gene panels to streamline diagnostic genomics. While achieving considerable success with a diagnostic yield slightly below 50%, an inventive proposal has emerged. The aim is to expand genetic testing boundaries by improving.ariant identification and classification and synergistically integrating genomic technologies with clinical expertise. This ambitious approach aims to enhance genetic diagnosis standards and improve the application of clinical best practices, representing a significant step forward in comprehensive patient renal care
Hypothesis and aimsOur overarching hypothesis is that the diagnostic yield for patients with suspected genetic kidney disease can be improved by combining the re-analysis of existing genomic data with advanced sequencing and analysis techniques and functional genomics approaches. To test this hypothesis, we have developed the following research aims:
Aim 1. Patient IdentificationThis aim centres around identifying individuals more likely to have a monogenic cause of their CKD through a targeted recruitment survey and specific criteria. We will focus on individuals within the KidGen Collaborative KGC network who remain undiagnosed or have inconclusive results despite undergoing targeted genetic testing, clinical WES, or clinical WGS.
Aim 2. Enhanced testing approachThis aim has a two-fold approach:
1.Re-analysis of existing genomic data. We will re-analyse existing genomic data from patients recruited to previous KidGen studies who remain without a genetic diagnosis.
2.Advanced genomic testing. We will conduct advanced genomic testing for existing compelling cases where the initial re-analysis did not yield conclusive results or for prospective undiagnosed cases after clinical diagnostic testing.
Aim 3. Functional modelling and validationThis aim focuses on compelling VUS in known and newly identified genes associated with kidney phenotypes. Where suitable, we will use CRISPR/Cas9 gene editing technology to introduce VUS into the mouse genome to study their impact on kidney function. Again, where appropriate, we will also use patient-derived and other genetically engineered induced pluripotent stem cells (iPSCs) to create kidney organoids and assess the contribution of VUS to kidney disease phenotypes [16,17,18]. Additionally, we will validate the functional impact of those VUS suspected to alter gene splicing. The primary goal of this aim is to provide additional evidence of the association between specific genetic variants and kidney disease phenotype. This will include VUS and new gene-disease associations. The objective will be to generate sufficient evidence to reclassify VUS as pathogenic or likely pathogenic or establish a novel gene-disease association.
Aim 4. Establishment of KidGen databasesThis aim centres around establishing a robust participant database. This database will include patient demographics, clinical characteristics, and genetic profiles. We aim to create a comprehensive database within kidney genetics research for multiple purposes. This database will primarily guide phenotype-driven variant interpretation and sequencing strategies for the current study. In addition, the data collected will inform recruitment strategies for future studies, such as clinical trials for new treatments. The database will also assist with implementation science, health economics, and quality assurance efforts by providing insights into patient populations and their specific needs and assessing equity of access.
Methodology—Aim 1. Patient identificationTo achieve the objectives outlined in Aim 1, our recruitment strategy involves enrolling up to 200 and their relevant family members. Of these, 100 patients will originate from established Australian Genomics kidney cohorts, including the KidGen Collaborative Kidney Genetics Cohort [19] and the HIDDEN (wHole genome Investigation to iDentify unDEtected Nephropathies) Cohort [20] families who have previously undergone genomic testing for their kidney condition but remain undiagnosed. These patients have already provided informed consent for subsequent analyses during their initial enrolment. In addition to these existing cohorts, we plan to prospectively enrol up to 100 new patients and relevant family members through KGCs between mid-2022 and mid-2026. Individuals will be selected based on stringent inclusion criteria, including a substantial likelihood of a monogenic cause for their GKD despite previous uninformative genetic or genomic testing (Table 1). Informed consent can be provided by paper or electronic consent via platforms such as Research Electronic Data Capture (REDCap) or CTRL (Control) instruments [21, 22].
Table 1 KidGen study participation criteria: inclusion and exclusionThe KidGen Collaborative comprises a network of twenty KGCs across Australia. These clinics will serve as recruitment sites and are geographically distributed as follows: four in New South Wales (NSW), one in the Northern Territory (NT), three in Queensland (QLD), two in South Australia (SA), one in Tasmania (TAS), five in Victoria (VIC), and four in Western Australia (WA) as detailed on Fig. 2.
Fig. 2
Australia's renal genetic services: a geographic breakdown. The KidGen Collaborative includes a network of 19 adult (green pins) and pediatric (blue pins) kidney genetics clinics (KGCs) operating nationwide. KGCs receive diagnostic support from the National Association of Testing Authorities (NATA)-accredited labs (orange pins), while research groups (purple pins) conduct functional genomics activities and perform subsequent variant curation in the background. NT, Northern Territory; QLD, Queensland; NSW, New South Wales; ACT, Australian Capital Territory; VIC, Victoria; TAS, Tasmania; SA, South Australia; WA, Western Australia
To be nominated by KGC, patients must meet all inclusion criteria and none of the exclusion criteria, as listed in Table 1. A rigorous selection process will be carried out by the Clinical Working Group (CWG) using a phenotype-driven approach [23]. The CWG reviews all patients nominated by the KGCs to ensure patients meet the selection criteria. Based on their evaluation, the CWG will recommend accepting or declining inclusion in the study. A majority of CWG members must recommend recruitment for the patient to be enrolled in the study. Where further discussion is beneficial, referring clinicians may be asked to present nominated cases at the monthly National Multidisciplinary Team (nMDT) meeting. A referral score will be calculated for each patient by evaluating five key domains based on evidence suggesting a high likelihood of monogenic kidney disease (Table 2). While this score is not the sole referral determinant, it serves as an empiric and objective metric for which diagnostic yield can be reported. High-scoring patients (≥ 10) are prioritised for direct admission to the study, while lower and low-scoring patients (≤ 6) may only be accepted by consensus agreement after presentation at the nMDT. Suggestions for refinement of this rubric will be made based on study outcomes.
Table 2 Experimental referral scoring systemMethodology—Aim 2. Enhanced testing approachThis study will use a standardised analysis pipeline using a computational tool, Hail Batch, to (re)process all short-read exome and genome data. Single nucleotide variants (SNVs) and small insertion/deletions (indel) will be called with GATK HaplotypeCaller, copy number variants (CNVs) and structural variants (SV) will be called with GATK-SV for WGS data [24] and GATK-gCNV for exome data [25]. An adapted version of the GATK mitochondrial variant calling pipeline will be used to call identical homoplasmic and heteroplasmic mixtures of variants in mtDNA from WGS data using the mitochondria mode of GATK MuTect2 [26]. Short tandem repeat expansions at known disease-associated loci will be evaluated with STRipy [27]. All variant data will be imported into the CPG-hosted instance of the seqr tool, a web-based analysis and collaboration tool, for curation and analysis [15].
Existing sequencing data from the retrospective cohort will undergo reanalysis using state-of-the-art analytical methods. Raw genomic data will be reprocessed using the standardised analysis pipeline [28] before reanalysis is performed using the automated reanalysis tool Talos [29] and manual curation by a variant analyst.
Prospectively recruited patients and those in the retrospective cohort who did not have suitable historical sequencing data will undergo re-sequencing. This will predominantly be based on short-read whole genome sequencing (short-read WGS). Long-read sequencing will be considered case-by-case for undiagnosed patients, mainly after negative WES analysis. This includes considering long-read WGS for patients with a broad phenotype and targeted (real-time selective sequencing) long-read sequencing for patients suspected of having specific genes or gene regions implicated in their disease. A dedicated variant analyst will lead the variant interpretation of short and long-read data. The variant analysis is performed iteratively with feedback from the referring clinician and clinical working group on the clinical relevance of identified variants. Research reports will be issued for newly identified genetic variants classified as “pathogenic” or “likely pathogenic,” following the guidelines of the American College of Medical Genetics and Genomics [30]. A highly suspicious VUS will undergo a functional genomic analysis to gain more pathological evidence.
Transcriptome sequencing (RNAseq) will be performed in specific cases where additional data are needed for interpretation or when investigating gene expression patterns (Fig. 3). Incorporating RNAseq allows for multimodal genomic profiling by combining different modalities of genomic data, such as short-read WGS/long-read WGS/RNAseq. Obtaining RNAseq data may require alternative sample types beyond DNA-based approaches. For example, skin fibroblast or existing tissue samples that were collected for clinical purposes but only partially utilised (e.g., kidney biopsy specimens). For patients with suspected splice-altering variants, we have developed collaborations with existing specialised groups to validate these variants further [31].
Fig. 3
KidGen research pathway for genomic testing. The foundation of KidGen’s genomic research is short-read whole genome sequencing (WGS) combined with advanced genomic analysis, a proven method for increasing diagnostic success. The process further involves in-depth genomic testing that prioritises variants based on patient traits and uses various advanced analysis methods, including functional genomics and long-read sequencing, and potentially integrates the results with gene expression data obtained from human urine-derived renal epithelial cells (HURECs). This process aims to provide information for diagnosis and potential discussions at a national multidisciplinary team (MDT) meeting to generate clinically reportable outcomes
Methodology—Aim 3. Functional modelling and validationParticipants with variants of uncertain significance, either due to an uncertain variant identified in an established kidney-disease gene or a variant identified in a gene with a novel or uncertain gene-disease association, will be discussed via the clinical and functional working groups, and when required, the nMDT. Consideration will be given to the appropriateness of animal or organoid modelling to establish a definitive diagnosis (Fig. 4).
Fig. 4
Mouse vs. organoid models: decoding kidney disease genetics. Mouse models and organoids serve as complementary tools in disease research, each offering distinct advantages. Mice are particularly well-suited for studying diseases caused by a small number of genes (candidate oligogenic genotypes), diseases affecting organs other than the kidneys (extra-renal phenotypes), and diseases with a later onset in life (adult-onset). Conversely, organoids excel in modelling diseases with early onset in life (antenatal/perinatal onset). They are valuable for developing personalised therapies because they can be cultured from a patient's cells
Induced pluripotent stem cell (iPSC) lines carrying patient VUS will be reprogrammed from patient-derived peripheral blood mononuclear cells or skin fibroblasts, with or without simultaneous gene correction of the VUS [32, 33]. In circumstances where patient cells are not easily accessible, patient VUS may be gene-edited into wild-type cell lines. iPSC will be differentiated into kidney organoids using established protocols [32, 34, 35]. Kidney organoids are stem cell-derived models of human kidney tissue containing multiple kidney cell types arranged into segmented in vitro nephron-like structures. Kidney organoids offer distinct advantages to the functional genomic modelling of human GKD, with important limitations including gene expression equivalent to second-trimester human foetal kidney and lack of tubular urine flow and vasculature in an in vitro setting, reviewed in [36]. Accordingly, VUS will be prioritised for modelling organoids based on candidate gene expression in transcriptional profiling organoid datasets and the expectation of an in vitro readout of disease. The organoid research will be conducted at the Murdoch Children's Research Institute (Melbourne, Australia), which is built on an established organoid-based functional genomic research program.
The second approach for functional assessment of VUS involves creating a mouse model with a VUS of interest identified in the patient. This is done by introducing the VUS into the mouse genome using CRISPR/Cas9 gene-editing technology. This enables subsequent phenotyping related to kidney function. Notably, there is no requirement for patient samples in these animal models. The goal is to enhance our understanding of the underlying mechanisms contributing to the patient's kidney disease. The CRISPR/Cas9 gene-editing work with mice will be conducted at the Monash Genome Modification Platform within Monash University (Melbourne, Australia). The expertise and facilities available at this institution ensure optimal conditions for conducting precise and controlled genetic manipulations [37].
Methodology—Aim 4. Establishment of KidGen databasesReferring physicians enter phenotypic data for all recruited patients into a specially designed REDCap database through targeted surveys structured to capture detailed patient phenotypes while minimising data entry burdens and ensuring data completeness.
We aim to create a centralised, de-identified repository within the KidGen database, housing clinical, demographic, original and de novo sequencing data and re-analysis results. By consolidating these resources on a single platform, the initiative provides streamlined access for KidGen researchers and collaborators, including those working in implementation science, health economics, and patient advocacy. It also strengthens data integrity and security through adherence to strict de-identification protocols. Most importantly, it ensures that critical research findings are readily available for multidisciplinary expert analysis, unlocking new insights into kidney genetics and fostering collaboration with far-reaching implications for patient care.
Ethical implications and considerationsEthical considerations are paramount in the KidGen National Kidney Genomics Study. All participants will provide informed consent via paper or electronic platforms like REDCap or CTRL, ensuring they understand the study's aims, procedures, potential risks, and benefits. Data privacy will be maintained through strict adherence to national and institutional guidelines, including data de-identification before analysis and storage in secure, password-protected REDCap databases hosted by the Murdoch Children's Research Institute (MCRI). Access to data will be restricted to authorised research personnel only. Furthermore, the study has received ethical approval (see Declarations), ensuring that all procedures are conducted ethically and in accordance with established standards. These measures are in place to safeguard participant confidentiality and ensure the responsible conduct of this research.
留言 (0)