
記住我

Graphical Abstract.
1 IntroductionHeart failure (HF) is the terminal stage of the progression of diverse functional or organic cardiovascular diseases, characterized by impaired ventricular filling and ejection capacity, with prevalent risk factors including hypertension, myocardial infarction, and myocardial disease (1). Epidemiological studies have indicated a global total of > 64 million patients with HF, with a prevailing trend toward a younger age of onset (2, 3). HF has emerged as a significant public health issue, posing risks to human health and escalating societal burden (2). Currently, the standard pharmacological interventions for HF management include angiotensin receptor-neprilysin inhibitors, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, sodium-glucose co-transporter 2 inhibitors, beta blockers, aldosterone receptor antagonists, and diuretics (4). Although they ameliorate HF to a certain extent, uncertainties persist concerning their long-term effects, and the potential adverse events associated with long-term medication are concerning. Therefore, there is an urgent need to develop safe and effective therapeutic strategies. HF is a clinical syndrome encompassing a range of complex pathological processes, including myocardial inflammation, myocardial fibrosis, myocardial hypertrophy, impaired angiogenesis, abnormal cardiac electrical signal conduction, energy metabolism disorders, and abnormal cardiomyocyte apoptosis (5–9). The activation of the NOD-like receptor protein 3 (NLRP3) inflammasome plays a crucial role in driving these pathological changes (5–8, 10). As an intracellular multiprotein complex, persistent or excessive activation of the NLRP3 inflammasome serves as a critical driver of both the onset and progression of HF, with the extent of its activation being strongly correlated with disease severity and patient prognosis (5, 11, 12). Therefore, inhibiting the activation of the NLRP3 inflammasome holds promise as a novel breakthrough in the prevention and treatment of HF.
Traditional Chinese medicine (TCM) has been recognized as a promising therapeutic strategy for HF, owing to its ability to effectively reverse adverse cardiac remodeling, lower rehospitalization and mortality rates, and enhance the quality of life of patients (13, 14). The active ingredients of TCM are the material basis for its therapeutic effects and constitute the focal point of research on TCM. Relevant studies have indicated that the active ingredients of TCM can mitigate the onset and progression of HF by inhibiting the NLRP3 inflammasome (15–17). Thus, this study summarizes the role of NLRP3 inflammasome activation in the onset and progression of HF, as well as the current research on the use of TCM active ingredients to prevent and treat HF through targeted inhibition of the NLRP3 inflammasome, aiming to provide insights for future basic research and novel drug development.
2 NLRP3 inflammasome2.1 Structure of the NLRP3 inflammasomeThe innate immune system is the first line of defense in the human body. Innate immune cells activate inflammasome by recognizing pathogen-associated molecular patterns and damage-associated molecular patterns via pattern recognition receptors, subsequently initiating inflammatory responses (12). The NLRP3 inflammasome is the most widely and intensively studied inflammasome and is a multiprotein complex comprising the NLRP3 protein, apoptosis speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 precursor (pro-caspase-1) (12) (Figure 1). NLRP3 acts as a sensor and consists of a central NACHT domain, a leucine-rich repeat (LRR) domain at the carboxyl-terminal (C-terminal), and a pyrin domain (PYD) at the amino-terminal (N-terminal). The NACHT domain primarily facilitates NLRP3 protein oligomerization and contains an adenosine triphosphatase active site, enabling the regulation of NLRP3 protein activity through adenosine triphosphate (ATP) hydrolysis (18). The LRR domain mediates protein–protein interactions and plays a crucial role in NLRP3 inflammatory signaling by recognizing and interacting with both exogenous and endogenous molecules (19). PYD recruits downstream effector signaling molecules that trigger inflammasome assembly (20). ASC functions as an adaptor with two domains: the N-terminal PYD and the C-terminal caspase activation and recruitment domain (CARD) (21). The PYD of ASC corresponds to the homotypic PYD of NLRP3 proteins, which mediates the interaction between ASC and NLRP3 proteins (20, 21). The CARD of ASC is responsible for binding pro-caspase-1 (21). Pro-caspase-1 functions as an effector and comprises three domains: the N-terminal CARD, central large catalytic subunit domain p20, and C-terminal small catalytic subunit domain p10 (22). The CARD of pro-caspase-1 is responsible for interactions with the CARD of ASC (22). Subsequently, p20 and p10 facilitate the cleavage of the interleukin-1β precursor (pro-IL-1β) and interleukin-18 precursor (pro-IL-18) into mature forms of IL-1β and IL-18.
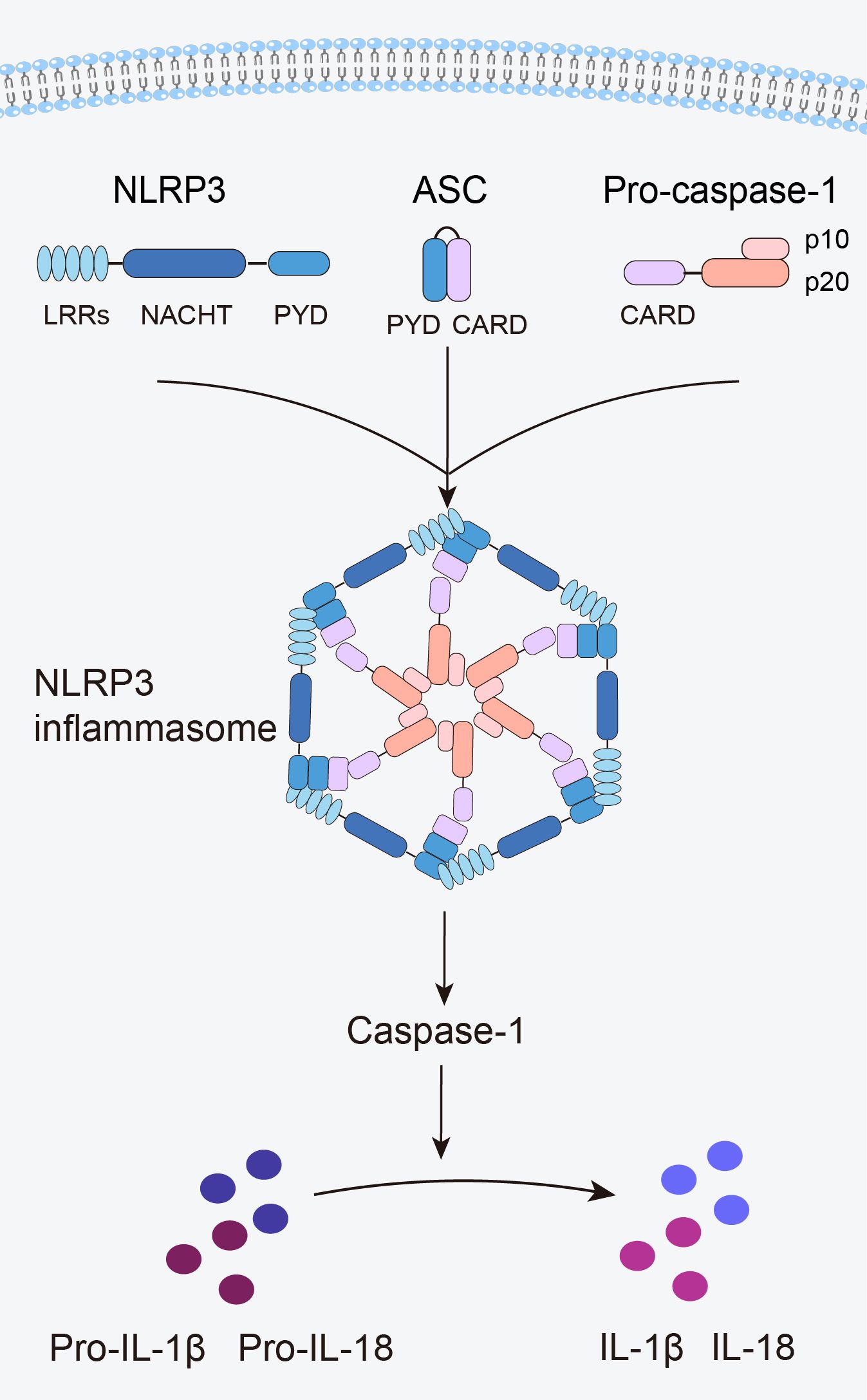
Figure 1. Structure of the NLRP3 inflammasome.
When an organism encounters an external stimulus, NLRP3 interacts with the PYD of ASC via its PYD. The CARD of ASC recruits and binds to the CARD of pro-caspase-1, triggering self-cleavage of pro-caspase-1 to yield active caspase-1. Caspase-1 cleaves pro-IL-1β and pro-IL-18 to generate mature IL-1β and IL-18, thus initiating an inflammatory response (Figure 1).
2.2 Mechanism of NLRP3 inflammasome activationThree distinct pathways exist for NLRP3 inflammasome activation: canonical NLRP3, non-canonical NLRP3, and alternative NLRP3 inflammasome activation (23) (Figure 2).
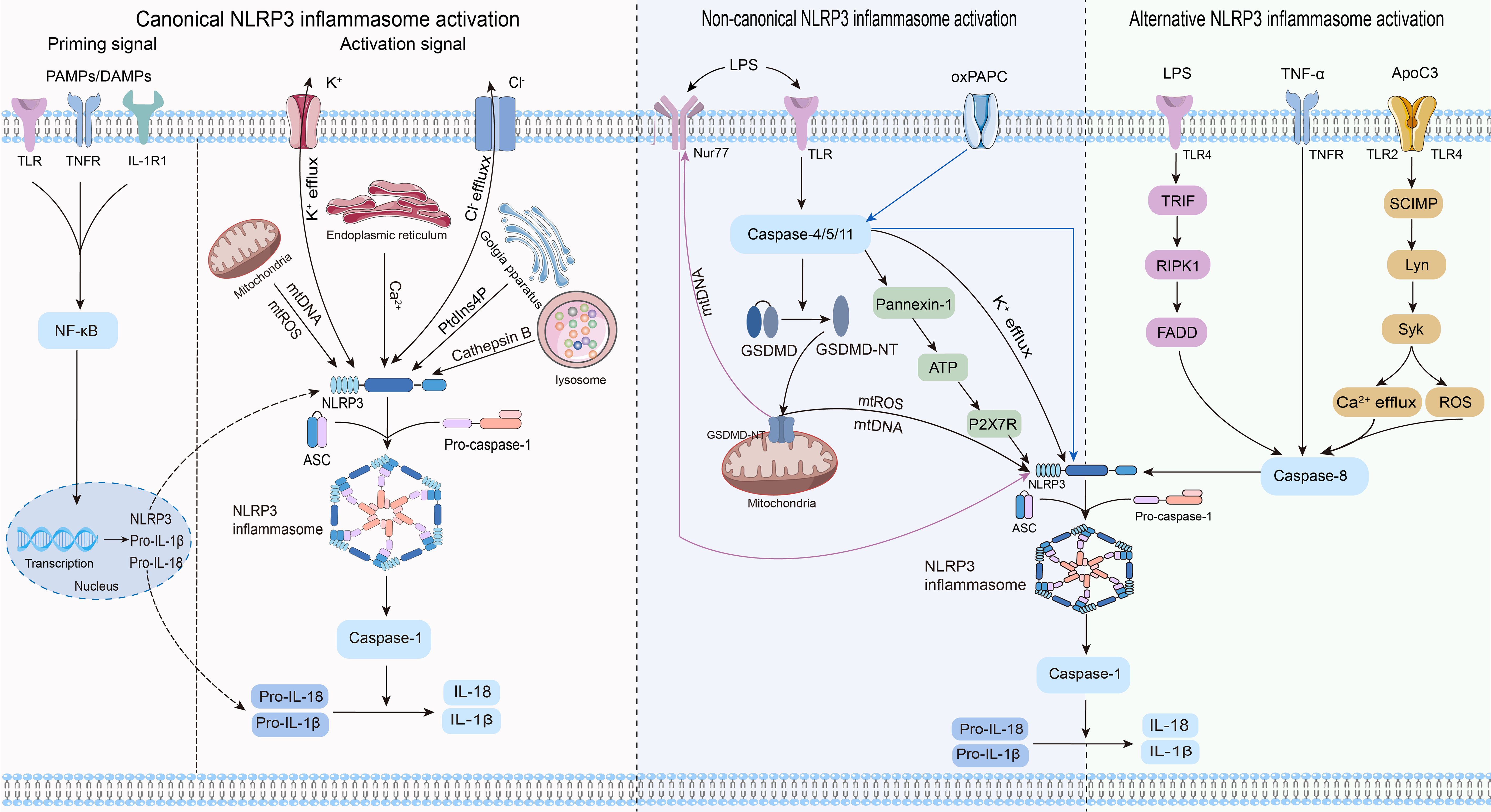
Figure 2. Three pathways and related mechanisms of NLRP3 inflammasome activation.
2.2.1 Canonical NLRP3 inflammasome activationCanonical activation of the NLRP3 inflammasome involves two distinct processes: priming and activation (24). The priming phase involves transcriptional regulation and posttranslational modifications of NLRP3. Recognition of pathogen-associated molecular patterns or damage-associated molecular patterns by the corresponding pattern recognition receptors (25–27) triggers nuclear factor-κB (NF-κB) translocation and transcription, leading to increased expression of NLRP3, pro-IL-1β, and pro-IL-18 within the nucleus. Furthermore, priming signals trigger posttranslational modifications of NLRP3, including phosphorylation (28), ubiquitination (29), alkylation (30), S-nitrosylation (31), acetylation (32), and sumoylation (33), all of which are crucial for modulating the activation or inhibition of NLRP3. During the activation phase, NLRP3 responds to activating stimuli, subsequently initiating the assembly of the NLRP3 inflammasome, activation of caspase-1, and processing pro-IL-1β and pro-IL-18. This process ultimately produces proinflammatory cytokines IL-1β and IL-18, which are subsequently secreted into the extracellular space to trigger an inflammatory response.
Previous studies agree that the stimulus signals for NLRP3 inflammasome activation include potassium ion (K+) efflux (34), chloride ion (Cl-) efflux (35), mitochondrial dysfunction (36, 37), endoplasmic reticulum stress (38), trans-Golgi network catabolism (39), and the release of tissue protease B from damaged lysosomes (40). Remarkably, the interplay between some of these stimuli complicates the activation phase of NLRP3 (41).
2.2.2 Non-canonical NLRP3 inflammasome activationNon-canonical activation of the NLRP3 inflammasome primarily relies on the mediation of human caspase-4/5 or mouse caspase-11. CARDs of caspase-4/5/11 directly recognize lipopolysaccharides (LPS) from gram-negative bacteria, prompting their oligomerization of caspase-4/5/11 (42–44). This leads to the cleavage of GSDMD into its active form, GSDMD-NT, which in turn induces pyroptosis by creating pores in the cytoplasmic membrane (42–44). Notably, while inducing pyroptosis, caspase-4/5/11 do not directly cleave pro-IL-1β (45, 46). They are required to indirectly promote the cleavage of pro-IL-1β and the release of IL-1β through NLRP3-dependent activation of caspase-1 (45, 46). Research has revealed that caspase-4/5/11 can trigger the release of mitochondrial reactive oxygen species (mtROS) and mitochondrial DNA (mtDNA) by enhancing the pore-forming capability of GSDMD in the mitochondria, thereby contributing to the activation of the NLRP3 inflammasome (47, 48). The orphan receptor Nur77 is activated upon binding to LPS and mtDNA (48). Subsequently, Nur77 interacts with NLRP3, triggering activation of the NLRP3 inflammasome (48). After LPS stimulation, caspase-4/5/11 trigger intracellular K+ efflux, one of the pathways by which caspase-4/5/11 mediates NLRP3 inflammasome activation (46, 49). Furthermore, the activated caspase-11 triggers the cleavage of pannexin-1 channels, leading to the release of ATP into the extracellular environment (50). Subsequently, the P2X7 receptor (P2X7R) responds to extracellular ATP, triggering the assembly of NLRP3 inflammasome and the release of IL-1β (50). The scaffold structural domain of pro-caspase-11 facilitates the activation of NLRP3 through interaction with the LRRs and PYD of NLRP3 (51). Intriguingly, this activation is mediated by the co-induction of live gram-negative bacterial mRNA and LPS (51). In addition to LPS, oxidized phospholipids (oxPAPC) serve as endogenous ligands for caspase-11 (52). It induces the oligomerization of caspase-11 by binding to its catalytic domain in dendritic cells, thereby promoting the assembly of the NLRP3 inflammasome and inflammation (52). Nevertheless, indications suggest that oxPAPC exerts an anti-inflammatory effect as it can diminish the non-canonical activation of the macrophage NLRP3 inflammasome and dampen the inflammatory response through competitive binding of LPS to caspase-4 and caspase-11 (53). Consequently, further investigations are warranted to explore the potential divergent effects of oxPAPCs on non-canonical NLRP3 inflammasome activation across different cell types.
2.2.3 Alternative NLRP3 inflammasome activationIn contrast to the previously mentioned activation pathways, the alternative activation pathway requires only one step to activate the NLRP3 inflammasome and lacks the features of canonical and non-canonical NLRP3 inflammasome activation, such as K+ efflux, pyroptosis, or pyroptosome formation (54). This activation pathway exhibits species specificity and has been identified exclusively in human and porcine monocytes (54). Research revealed that toll-like receptor (TLR) 4 in human monocytes triggered the NLRP3 inflammasome through the TRIF/RIPK1/FADD/caspase-8 signaling pathway upon stimulation by LPS, eliminating the need for a secondary signal for mediation (54). Tumor necrosis factor-α (TNF-α), a closely associated cytokine in psoriasis, selectively induces the initiation of the NLRP3 inflammasome through the TNFR/caspase-8 pathway even without an initial signal (55). Apolipoprotein C3 (ApoC3), an endogenous mediator, selectively triggers activation of the NLRP3 inflammasome in human monocytes (56). This process involves the formation of heterodimers between TLR2 and TLR4, initiating a pathway dependent on SCIMP/Lyn/Syk for calcium influx and ROS production, leading to caspase-8 activation and ultimately triggering activation of the NLRP3 inflammasome (56).
3 Role of NLRP3 inflammasome activation in the onset and progression of HFUpon systematically reviewing studies on NLRP3 inflammasome activation in HF, we discovered that its activation promotes the onset and progression of HF by exacerbating multiple crucial pathophysiological processes. These pathological changes include myocardial inflammatory injury, adverse myocardial fibrosis, pathological myocardial hypertrophy, inhibited angiogenesis, abnormal cardiac electrical signal conduction, disturbed cardiac energy metabolism, and abnormal cardiomyocyte apoptosis (Table 1). Among these processes, myocardial inflammatory injury stands as a central nexus, where chronic inflammation not only directly harms the myocardium but also has the potential to initiate a cascade of events that worsen other pathological alterations in HF. Notably, the activation of the NLRP3 inflammasome involves the modulation of multiple signaling pathways, which are pivotal in mediating the aforementioned pathological processes (Figure 3).
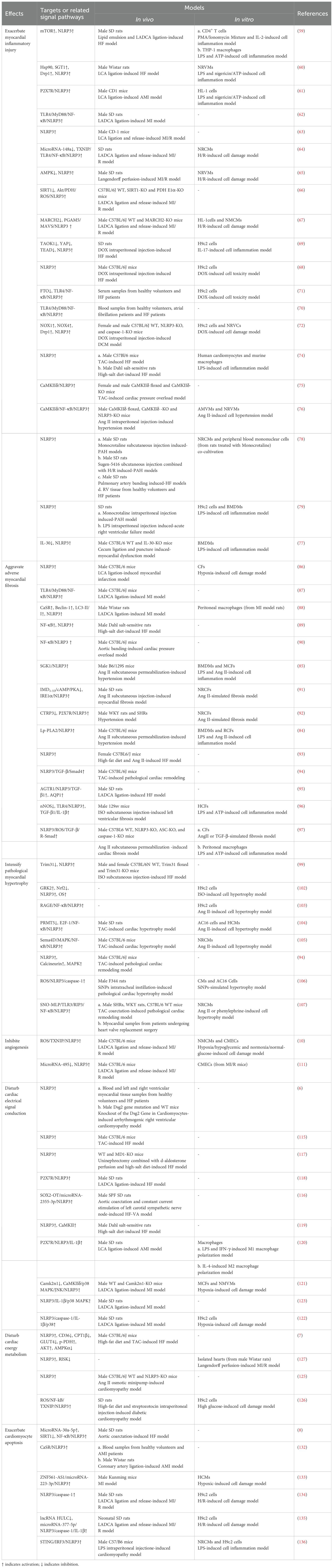
Table 1. Role of NLRP3 inflammasome activation in the onset and progression of HF.
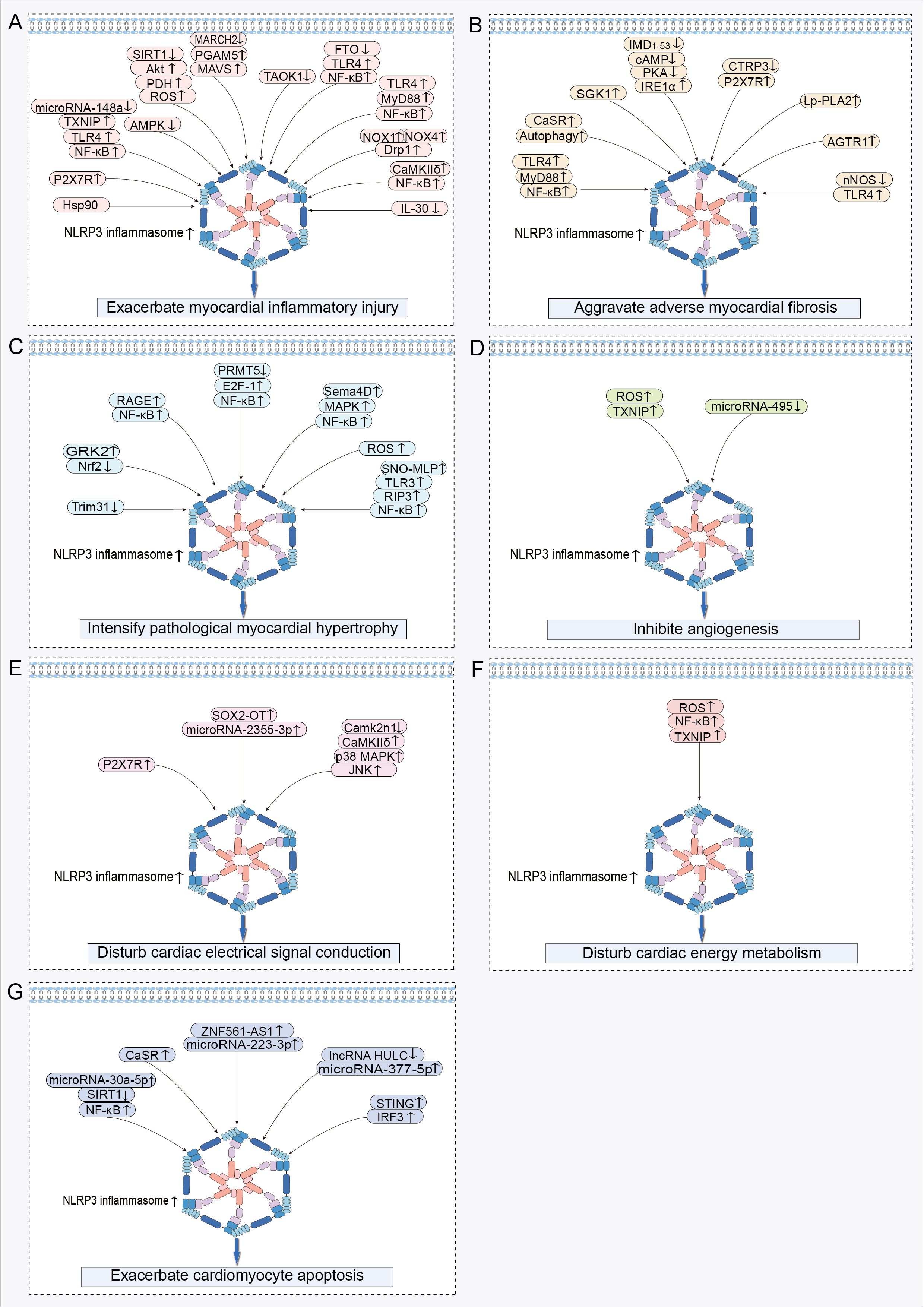
Figure 3. Signaling pathways regulating NLRP3 inflammasome activation in HF: (A) Signaling pathway exacerbating myocardial inflammatory injury. (B) Signaling pathway aggravating adverse myocardial fibrosis. (C) Signaling pathway intensifying pathological myocardial hypertrophy. (D) Signaling pathway inhibiting angiogenesis. (E) Signaling pathway disturbing cardiac electrical signal conduction. (F) Signaling pathway disturbing cardiac energy metabolism. (G) Signaling pathway exacerbating cardiomyocyte apoptosis. ↑ indicates activation; ↓ indicates inhibition.
3.1 Exacerbate myocardial inflammatory injuryAn appropriate inflammatory response serves as a protective mechanism that eliminates harmful stimuli and repairs damaged tissues (57). However, excessive or prolonged inflammation escalates the risk of cardiac dysfunction and adverse cardiac remodeling (57, 58).
In the cardiac tissues of HF rats, increased NLRP3-positive spots, caspase-1 shear activation, and elevated levels of mature IL-1β were accompanied by a heightened inflammatory response (59, 60). These findings suggest that the NLRP3 inflammasome plays a contributory role in the development of myocardial inflammation in HF (59, 60). During acute myocardial infarction (AMI), dying heart myocytes initiate the assembly of the NLRP3 inflammasome by activating P2X7R via ATP release (61). This process amplifies cardiac inflammation, leads to further loss of functional myocardium, and even results in HF (61). Following myocardial infarction (MI), myocardial injury triggers the activation of the NLRP3 inflammasome, which exacerbates the myocardial inflammatory response, leading to enlargement of the infarct and worsening of cardiac dysfunction (62). Nicorandil pretreatment decreased NLRP3 inflammasome activation by inhibiting the TLR4/myeloid differentiation primary response protein 88 (MyD88)/NF-κB pathway, thereby alleviating the detrimental effects of MI on the heart (62). Activation of the NLRP3 inflammasome plays a crucial role in promoting myocardial ischemia/reperfusion (MI/R) injury (63). MI/R injury results in decreased microRNA-148a expression in myocardial cells, which increases the expression of thioredoxin-interacting protein (TXNIP) (64). Subsequently, TXNIP activates the TLR4/NF-κB/NLRP3 signaling pathway, promoting the release of inflammatory factors IL-1β and IL-18, thereby increasing inflammatory cell death in myocardial cells, leading to more extensive myocardial damage and worsening of cardiac function (64). Furthermore, studies have revealed that signaling pathways including adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) (65), silent information regulator of transcription (SIRT) 1/serine/threonine protein kinase B (Akt)/pyruvate dehydrogenase (PDH)/ROS (66), and E3 ubiquitin ligase membrane-associated RING finger protein 2 (MARCH2)/phosphoglycerate mutase 5 (PGAM5)/mitochondrial anti-viral-signaling protein (MAVS) (67) contribute to the exacerbation of cardiac inflammatory injury by activating the NLRP3 inflammasome, thereby exacerbating adverse cardiac outcomes caused by MI/R.
Activation of the NLRP3 inflammasome is a pivotal factor contributing to increased inflammatory damage in the non-ischemic myocardium. Activation of the NLRP3 inflammasome led to increased cardiomyocyte pyroptosis and reduced proliferative capacity in the doxorubicin (DOX)-induced HF model, collectively exacerbating the pathogenic progression of HF (68, 69). Mechanistically, activation of the TLR4/MyD88/NF-κB signaling pathway serves as an upstream event that triggers the activation of the NLRP3 inflammasome, thereby exacerbating cardiomyocyte pyroptosis and myocardial inflammation in DOX-induced HF (70, 71). Myocardial tissues from patients with dilated cardiomyopathy (DCM) exhibit aberrant NLRP3 inflammasome activation and pronounced pyroptosis, which are correlated with diminished cardiac function (72). In a DOX-induced DCM mouse model, DOX triggered the hyperactivation of the NLRP3 inflammasome by upregulating NOX1 and NOX4 expression and activating dynamin-related protein 1 (Drp1)-dependent mitochondrial fragmentation (72). This process exacerbates cardiomyocyte pyroptosis and contributes to the progression of cardiac dysfunction (72). Macrophages play a pivotal role in the regulation of cardiac inflammation (73). In the HF state, the activation of the NLRP3 inflammasome in myocardial tissue promotes macrophage infiltration into the heart (74). Mechanistic study has demonstrated that cardiomyocytes activate NLRP3 through the calmodulin-regulated kinase δ (CaMKIIδ) signaling pathway, promoting the release of pro-inflammatory cytokines IL-1β, IL-18, and IL-6, as well as the production of monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein 1α (75). These factors synergistically promote macrophage migration to myocardial tissue, thereby further amplifying cardiac inflammation (75). Furthermore, activation of myocardium-specific CaMKIIδ can also mediate the activation of the NLRP3 inflammasome through the NF-κB pathway, leading to increased macrophage recruitment to the damaged myocardium and exacerbating the cardiac inflammatory cascade (76). Interestingly, macrophages demonstrate two pro-inflammatory effects, pyroptosis and pro-inflammatory polarization, upon recruitment to the heart (77–79). In pulmonary arterial hypertension (PAH)-induced right ventricular failure, there was a significant increase in the number of macrophages within the right ventricle, accompanied by an elevated expression of the NLRP3 inflammasome in these macrophages (78). The elevated expression of the NLRP3 inflammasome not only promoted macrophage pyroptosis, but also drove macrophages toward a pro-inflammatory M1-type phenotype (78). This shift exacerbated the inflammatory response in the right ventricle, contributing to further deterioration of right ventricular dysfunction (78). Additionally, cardiomyocyte NLRP3-dependent pyroptosis further stimulates macrophage polarization toward a pro-inflammatory M1 phenotype in myocardial tissues through the release of pro-inflammatory cytokines and MCP-1 (79). Similarly, in sepsis-induced cardiac inflammatory injury and dysfunction, macrophage pyroptosis in cardiac tissues, along with the polarization of Ly6Chigh macrophages, is positively regulated by NLRP3 complex activation (77). These studies confirm that activation of the NLRP3 inflammasome exacerbates the cardiac inflammatory cascade by promoting macrophage recruitment to the heart and stimulating macrophage pyroptosis and pro-inflammatory polarization.
3.2 Aggravate adverse myocardial fibrosisMyocardial fibrosis is characterized by abnormal proliferation and differentiation of cardiac fibroblasts (CFs) and excessive accumulation and abnormal distribution of the extracellular matrix (ECM). Myocardial fibrosis is a critical reparative response aimed at maintaining cardiac integrity after myocardial injury (80). However, excessive myocardial fibrosis results in diminished myocardial compliance and cardiac diastolic and systolic dysfunction, serving as a pivotal pathological foundation for the onset and progression of HF (81, 82).
Myofibroblasts play a crucial role as mediator cells in the progression of myocardial fibrosis (83). They induce cardiac fibrous scar formation and dysfunction by synthesizing significant quantities of ECM and collagen, secreting pro-fibrotic cytokines, and expressing α-smooth muscle actin (α-SMA) (83). IL-1β was identified as a key mediator in promoting the proliferation and differentiation of CFs into myofibroblasts, indicating that NLRP3 inflammasome activation is an important factor mediating the progression of myocardial fibrosis (84, 85). MI leads to a notable upregulation in the expression of fibrotic markers in myocardial tissues, including collagen I, collagen III, and α-SMA (86). Importantly, the degree of NLRP3 inflammasome activation is positively correlated with the severity of myocardial fibrosis (86). Myocardial ischemia triggered the activation of the TLR4/MyD88/NF-κB signaling pathway, which facilitated the assembly and activation of the NLRP3 inflammasome, thereby exacerbating cardiac inflammation (87). The progression of inflammation enhances fibrosis, resulting in increased cardiac stiffness and reduced cardiac pumping function (87). The expression of calcium-sensitive receptor (CaSR) is elevated in myocardial tissue following MI (88). CaSR exacerbates both inflammation and fibrosis post-MI by activating the autophagy/NLRP3 inflammasome pathway (88).
In pressure overload-induced HF, the activation of the NLRP3 inflammasome was identified as a critical factor driving the progression of myocardial fibrosis (89). Mechanistically, chronic stress overload initiates the activation of the NF-κB/NLRP3 inflammasome pathway (90). This pathway amplifies the aberrant activation of cardiac fibroblasts and promotes the over-synthesis of collagen associated with fibrosis, thereby fueling the adverse progression of cardiac fibrosis (90). Administration of angiotensin II (Ang II) induced myocardial fibrosis in mice, as indicated by the excessive deposition of collagen fibers, elevated expression levels of transforming growth factor-β (TGF-β) and connective tissue growth factor, along with NLRP3 inflammasome activation in cardiac tissues and heightened IL-1β secretion (85). Notably, treatment with MCC950 successfully reversed these pathological alterations (85). Inositol-requiring enzyme 1α (IRE1α) acted as a sensor of endoplasmic reticulum stress, capable of triggering NLRP3 inflammasome activation, thereby exacerbating the progression of myocardial fibrosis (91). The endogenous cardiovascular protective peptide, intermedin1-53 (IMD1-53), had the ability to reduce the expression of IRE1α through the activation of the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathway, leading to the inhibition of NLRP3 inflammasome activation and mitigation of Ang II-induced cardiac fibrosis (91). Reduced expression of C1q/TNF-related protein-3 (CTRP3) in myocardial tissue is linked to the advancement of cardiac fibrosis (92). The restoration of CTRP3 expression ameliorated Ang II-induced myocardial fibrosis by inhibiting the P2X7R/NLRP3 inflammasome pathway to reduce α-SMA, collagen I/III, and matrix metallopeptidase (MMP) 2/9 expression (92).
TGF-β is a central signaling pathway in the promotion of fibrosis. In HF, activation of the NLRP3 inflammasome is considered a significant factor in the upregulation of TGF-β gene expression in the cardiac tissue (93). Specifically, the activated NLRP3 inflammasome promotes the advancement of cardiac fibrosis by triggering the TGF-β/Smad4 signaling pathway to enhance the expression levels of collagen type I, collagen type III, MMP-2, MMP-9, and α-SMA (94). Further study has demonstrated that Ang II receptor type 1 (AGTR1) accelerates myocardial fibrosis progression by activating the NLRP3 inflammasome and enhancing the production of TGF-β1 (95). Concurrently, activation of the TLR4 receptor also triggers NLRP3 inflammasome activation, initiating a signaling cascade that enhances the pro-fibrotic effects of the TGF-β1/IL-1β axis, promotes cardiac myofibroblast differentiation, increases interstitial collagen deposition, and ultimately exacerbates fibrosis (96). Additionally, a study revealed the mitochondrial localization of NLRP3 in CFs and demonstrated that NLRP3 is involved in the development of cardiac fibrosis by enhancing mitochondrial ROS production, promoting activation of the TGF-β/R-Smad pathway, and facilitating CF differentiation (97).
3.3 Intensify pathological myocardial hypertrophyPathological myocardial hypertrophy is an adaptive response of the heart to prolonged pressure or increased volume overload. Nevertheless, as myocardial hypertrophy advances to a certain level, it can exert significant adverse effects on the cardiac structure and function, thereby increasing the risk of HF (98).
In the context of cardiac remodeling, the activation of the NLRP3 inflammasome promotes not only cardiac inflammation and fibrosis but also aggravates pathological myocardial hypertrophy, consequently exacerbating symptoms of HF (99). Elevated levels of G protein-coupled receptor kinase 2 (GRK2) were identified in hypertrophied myocardial tissue (100, 101). Mechanistically, GRK2 promotes the activation of the NLRP3 inflammasome and induces oxidative stress (OS) by downregulating the expression of nuclear factor erythroid-2-related factor 2 (Nrf2), thereby exacerbating isoproterenol (ISO)-induced pathological cardiac hypertrophy (102). The receptor for advanced glycation endproducts (RAGE) participated in Ang II-induced pathological cardiomyocyte hypertrophy by activating the NF-κB/NLRP3/IL-1β pathway (103). Under pressure overload, there is a reduction in protein arginine methyltransferase 5 (PRMT5) expression in hypertrophic myocardial tissues. Low PRMT5 expression triggered the activation of the E2F-1/NF-κB signaling pathway, leading to the activation of the NLRP3 inflammasome that promotes maladaptive cardiac hypertrophy induced by transverse aortic constriction (TAC) or Ang II (104). The overexpression of Sema4D contributed to pressure overload-induced cardiac hypertrophy (105). It promotes the assembly and activation of NLRP3 complexes by activating the mitogen-activated protein kinase (MAPK)/NF-κB signaling pathway, thereby exacerbating TAC-induced pathological cardiac hypertrophy and dysfunction (105). Elevated levels of calcineurin and MAPK phosphorylation were observed in the TAC surgery group of pressure-overloaded mice (94). In contrast, MCC950 ameliorates pathological cardiac hypertrophy and enhances cardiac function by inhibiting calcineurin expression and the MAPK signaling pathway (94). Silica nanoparticles (SiNPs) contributed to the exacerbation of cardiac hypertrophy (106). SiNPs worsen myocardial hypertrophy by inducing cardiomyocyte pyroptosis via activation of the ROS/NLRP3/caspase-1 signaling pathway (106). Transfection of cardiomyocytes with si-NLRP3 or the caspase-1 inhibitor VX-765 limited SiNP-induced pathological cardiac hypertrophy (106). S-nitrosylated muscle LIM protein (SNO-MLP) expression is markedly elevated in patients and animals with myocardial hypertrophy (107). This upregulation primarily facilitated the interaction between TLR3 and receptor-interacting protein kinase 3 (RIP3), thus initiating activation of the NF-κB/NLRP3 inflammasome pathway, ultimately fostering the progression of myocardial hypertrophy (107).
3.4 Inhibite angiogenesisAngiogenesis generates new blood vessels from existing capillaries or capillary post-veins. When the heart is exposed to ischemic and hypoxic stimuli, angiogenesis enhances its blood supply, thereby mitigating damage and preserving cardiac function resulting from ischemia and hypoxia (108, 109). However, the progression of cardiac pathological remodeling inhibits angiogenesis, resulting in decreased microvascular density and ultimately leading to HF (109).
Coronary microvessel rarefaction and decreased blood flow reserve have been identified as the primary drivers of diastolic dysfunction in patients with HF with a preserved ejection fraction (HFpEF) (9). Moreover, decreased cardiac microvascular density is intricately linked to NLRP3 inflammasome activation (110). Phosphorylation of microfibrillar-associated protein 4 (MAP4) downregulates the expression of angiogenic markers, such as CD31, CD34, VEGFA, VEGFR2, ANG2, and TIE2 (110). Mechanistically, MAP4 inhibited angiogenesis via NLRP3 inflammasome activation, leading to reduced cardiac microvessel density (110). Endothelial cells (ECs) serve as primary effector cells in cardiac angiogenesis, and any damage to or aberrant apoptosis of these cells significantly affects their capacity for cardiac angiogenesis. During MI/R injury, microvascular endothelial cells (CMECs) mediated interactions between TXNIP and NLRP3 by generating excessive ROS (10). This action subsequently escalates the activation level of the NLRP3 inflammasome, exacerbating damage to cardiac microvascular endothelial cells (10). In ischemia-reperfused myocardial tissues, there was a reduction in microRNA-495 expression, which facilitates the activation of the NLRP3 inflammasome, worsening inflammatory damage and apoptosis in CMECs (111). Conversely, elevating the expression of microRNA-495 or suppressing the NLRP3 gene decreases apoptosis and enhances the proliferation of CMECs by shifting the cell population from the G0/G1 phase to the S phase (111). This observation implies that the suppression of NLRP3 inflammasome activation may facilitate the repair and angiogenesis of cardiac microvessels. SIRT3 deficiency resulted in diminished expression of hypoxia-inducible factor-2α, VEGF, and angiopoietin-1, leading to decreased angiogenesis and subsequently causing coronary microvessel rarefaction and cardiac diastolic dysfunction (112). Trimethylamine N-oxide (TMAO) induces vascular inflammation by suppressing SIRT3 expression and superoxide dismutase 2 (SOD2) activity in endothelial cells, subsequently triggering mtROS/NLRP3 inflammasome signaling (113). Therefore, SIRT3 deficiency may impede coronary microvascular angiogenesis by activating the NLRP3 inflammasome.
3.5 Disturb cardiac electrical signal conductionVentricular arrhythmias (VAs) are common triggers and causes of death in HF (114). The cardiac electrical conduction system is crucial for maintaining normal heart function, and conduction abnormalities are the underlying precursors of arrhythmias.
Numerous studies have established that the activation of the NLRP3 inflammasome is a key factor in disrupting the electrical signaling in the heart and inducing malignant arrhythmias, particularly in the context of HF (6, 115–119). The activation of the NLRP3 inflammasome not only enhances myocardial inflammatory responses but also promotes the development of cardiac hypertrophy and fibrosis, creating a pro-arrhythmic environment (115). Additionally, NLRP3 inflammasome activation results in changes to myocyte ion channel functions, including a reduced expression of ion channel proteins such as Kv4.2, KChIP2, and Cav1.2, which affect the timing and morphology of cardiac action potentials and contribute to the development and maintenance of arrhythmias (115). Simultaneously, sympathetic nervous hyperactivity contributes to an increased susceptibility to HF-related ventricular arrhythmias due to NLRP3 inflammasome activation (117, 118). In particular, the activation of the NLRP3 inflammasome exacerbates cardiac sympathetic hyperactivity by promoting the release of the proinflammatory cytokine IL-1β, and this inflammatory-neural interaction results in altered electrophysiological properties of the heart, such as prolongation of the action potential duration and shortening of the effective refractory period, which increases the risk of ventricular arrhythmias (117, 118). In the myocardial tissues of rats with HF-related ventricular arrhythmias (VAs-HF), there was a notable increase in the expression of SOX2-overlapping transcript (SOX2-OT) and NLRP3 (116). Furthermore, silencing of the SOX2-OT gene reduced NLRP3 inflammasome activation levels by regulating microRNA-2355-3p, thus alleviating HF symptoms and diminishing VAs (116). In HFpEF, the activation of the NLRP3 inflammasome facilitates the development of atrial fibrillation through the promotion of atrial fibrosis, by prolonging the atrial action potential duration, increasing the dispersion of action potential duration, and activating inflammation-associated signaling pathways (119). Following MI, P2X7R facilitates the upregulation of nerve growth factor, tyrosine hydroxylase, and growth-associated protein 43 by mediating the activation of the NLRP3/IL-1β pathway, thereby fostering sympathetic sprouting (120). This cascade leads to altered cardiac electrophysiological characteristics and an increased susceptibility to arrhythmias (120). After MI, the expression of Camk2n1 is markedly reduced in the infarct border zone, leading to the activation of the CaMKIIδ/p38 MAPK/C-Jun N-terminal kinase (JNK)/NLRP3 inflammasome signaling pathway (121). This exacerbates myocardial fibrosis and increases the vulnerability to premature ventricular contractions, tachycardia, and ventricular fibrillation (121). Connexin 43 (Cx43) is a key regulator of cardiac electrical signal conduction (122, 123). The activation of the NLRP3 inflammasome within the myocardial infarct zone diminishes the expression of Cx43 in myocardial tissue, resulting in compromised intercellular communication and heightened vulnerability to VAs (122, 123). Conversely, restoring the expression of Cx43 in the heart by inhibiting the NLRP3/IL-1β/p38 MAPK pathway helps enhance cardiac conduction function and decrease the heart’s susceptibility to VAs (122, 123).
3.6 Disturb cardiac energy metabolismThe heart, as an organ with high energy and oxygen demands, relies on homeostasis of its energy metabolism as the foundational mechanism for maintaining the stability of the cardiac tissue structure and internal environment (124). The myocardial energy metabolism relies heavily on mitochondrial oxidative phosphorylation. When mitochondria are damaged, myocardial energy substrate utilization is altered, leading to decreased cardiac energy production and lactic acid build-up, which in turn affects cardiac energy metabolism and cardiomyocyte survival and accelerates the malignant progression of HF (124).
A complex interplay exists between NLRP3 inflammasome activation and myocardial energy metabolism disruption. Mitochondrial dysfunction is the trigger for the activation of the NLRP3 inflammasome (36, 37), while the activation of the NLRP3 inflammasome further impairs mitochondrial function and homeostasis (7, 125–127). In an obesity-associated HF model, overactivation of the NLRP3 inflammasome results in an imbalance between cardiac energy supply and demand, as evidenced by decreased fatty acid utilization and increased reliance on glycolysis and glucose oxidation in cardiomyocytes, thereby triggering cardiac metabolic reprogramming (7). This metabolic transition was concomitant with the downregulation of genes associated with mitochondrial energy transfer and respiratory pathways, consequently intensifying the advancement of HF (7). During MI/R injury, the inhibition of the NLRP3 inflammasome activates the reperfusion injury salvage kinase (RISK) pathway, subsequently enhancing the expression of markers associated with mitochondrial biogenesis and energy metabolism, such as mitochondrial transcription factor A, nuclear respiratory factor-1, and mitochondrial creatine kinase (127). These findings suggest an association between disturbed myocardial energy metabolism and the formation of the NLRP3 inflammasome complex during MI/R injury, indicating that inhibition of NLRP3 inflammasome activation contributes to the improvement of cardiac energy metabolism, thereby enhancing the resistance of cardiomyocytes to ischemic and hypoxic injury (127). In the Ang II-induced cardiomyopathy model, increased NLRP3 inflammasome activity was accompanied by decreased mtDNA copy number, reduced ATP synthase activity, increased ROS production, as well as mitochondrial structural alterations, including swelling, disordered matrix organization, and fragmentation (125). The knockdown of the NLRP3 gene mitigated Ang II-induced mitochondrial structural and functional damage, as well as alleviated cardiac dysfunction (125). In rats with diabetic cardiomyopathy, cardiomyocyte mitochondria exhibit swelling and matrix disorders, along with activation of the NLRP3 inflammasome (126). Silencing of the NLRP3 gene aided in restoring mitochondrial structure and reducing glycogenolysis and lipid accumulation in cardiomyocytes, suggesting an enhancement in cardiomyocyte energy metabolism (126).
3.7 Exacerbate cardiomyocyte apoptosisCardiomyocyte apoptosis is a type of programmed cell death that is genetically regulated (128). Cardiomyocytes, which are primary cardiac cells, are responsible for contraction (129). Excessive apoptosis of cardiomyocytes is a significant contributor to the structural alterations and functional deterioration of the heart. Moreover, it is a crucial driver of HF onset and progression (130, 131).
During the pathological progression of HF, the overactivation of NLRP3 exerts a pro-apoptotic effect on cardiomyocytes (8). Mechanistically, microRNA-30a-5p activates the NF-κB/NLRP3 signaling cascade by targeting SIRT1, thereby exacerbating cardiomyocyte apoptosis (8). CaSR expression is markedly elevated in the neutrophils of patients and rats with AMI (132). This upregulation facilitated NLRP3 inflammasome activation, release of IL-1β through the PLC-IP3 pathway, and calcium release from the endoplasmic reticulum (132). IL-1β interacted with the IL-1 receptor on cardiomyocytes, leading to an increase in Bax expression and caspase-3 cleavage, while decreasing Bcl2 expression, thereby effectively promoting cardiomyocyte apoptosis (132). In the myocardial tissue of MI mice, the expression of the long noncoding RNA zinc finger protein 561 antisense RNA 1 (ZNF561-AS1) is significantly upregulated (133). This upregulation leads to the inhibition of cardiomyocyte proliferation and augmentation of cardiomyocyte apoptosis via activation of the microRNA-223-3p/NLRP3 inflammasome pathway (133). During MI/R injury, activation of the NLRP3 inflammasome results in increased cardiomyocyte apoptosis through the upregulation of Bax protein expression and downregulation of Bcl2 expression (134). In myocardial tissues injured by ischemia reperfusion, the expression of long noncoding RNA highly up-regulated in liver cancer (lncRNA HULC) is downregulated (135). The decrease in lncRNA HULC expression results in heightened microRNA-377-5p activity, triggering the NLRP3/caspase-1/IL-1β signaling pathway (135). This cascade amplifies caspase-3 and cleaved-caspase-3 expression, ultimately worsening cardiomyocyte apoptosis (135). In a mouse model of cardiomyopathy, STING activation triggers the activation of the NLRP3 inflammasome by enhancing the phosphorylation and intranuclear translocation of IRF3 (136). This process elevates the ratios of Bax/Bcl-2 and C-Caspase3/T-Caspase3, leading to an increase in cardiomyocyte apoptosis (136).
4 TCM active ingredients in preventing and treating HF by inhibiting the NLRP3 inflammasomeActive ingredients are fundamental to the efficacy of TCM. Existing studies have revealed that active ingredients in TCM exert positive regulatory effects on key pathological processes of HF by inhibiting the NLRP3 inflammasome. In particular, these active ingredients are effective in ameliorating myocardial inflammation, adverse myocardial fibrosis, pathological myocardial hypertrophy, angiogenesis, cardiac electrical signal conduction, cardiac energy metabolism, and reducing abnormal cardiomyocyte apoptosis (Table 2). Further analysis revealed that these active ingredients, with the potential to prevent and treat HF, are primarily found in flavonoids and their glycosides, terpenes and their glycosides, phenolic acids, quinones, and phenylpropanoids (Table 2).
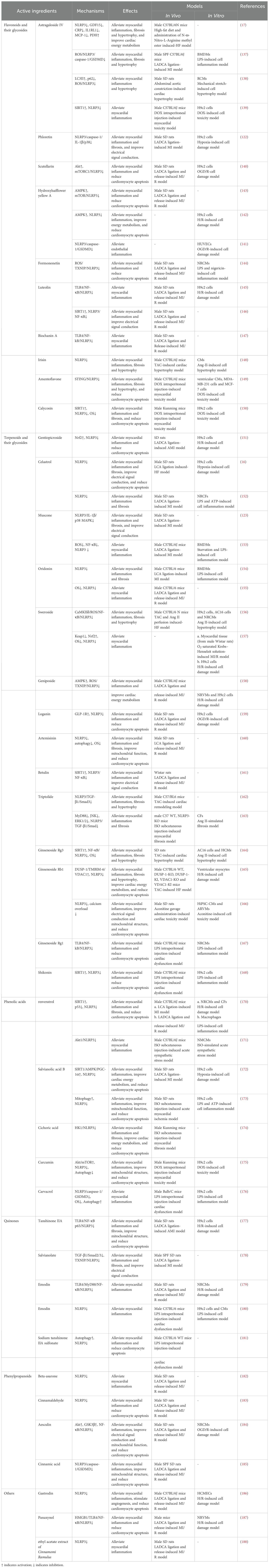
Table 2. Mechanisms of active ingredients in TCM regulating the NLRP3 inflammasome in HF.
4.1 Flavonoids and their glycosidesAstragaloside IV (AS-IV) demonstrates significant therapeutic potential for HFpEF (17). Specifically, AS-IV intervention markedly decreased NLRP3, IL-1β, and caspase-1 levels in the myocardium of HFpEF mice, with this reduction of biomarkers significantly linked to the amelioration of myocardial inflammation and enhancement of cardiac function (17). Additionally, AS-IV exerted a beneficial effect on maintaining cardiac metabolic homeostasis in HFpEF by optimizing cardiac glycolipid metabolism, enhancing mitochondrial function, and regulating energy metabolic pathways (17). AS-IV also effectively alleviated cardiac remodeling caused by MI (137). By inhibiting the ROS/caspase-1/GSDMD signaling pathway, As-IV reduces cardiomyocyte pyroptosis and lowers the expression levels of collagen I, collagen III, α-SMA, and fibronectin (137). This process effectively reduces post-MI cardiac fibrosis and hypertrophy, consequently enhancing the heart function (137). As-IV exerts protective effects against pressure overload-induced cardiac dysfunction (138). Through the upregulation of LC3II levels and inhibition of p62 expression, As-IV activated autophagy, subsequently inhibiting the ROS/NLRP3 inflammasome pathway and reducing the expression levels of IL-1β and IL-18 (138). This action effectively alleviates pressure overload-induced myocardial hypertrophy (138). In addition, As-IV mitigates DOX-induced myocardial toxicity (139). It exerts cardioprotective effects by reversing the DOX-induced downregulation of SIRT1 protein expression, upregulation of NLRP3 expression, and reduction in cardiomyocyte pyroptosis (139). Phloretin mitigates the electrical remodeling process in the heart post-MI (122). By inhibiting the NLRP3/caspase-1/IL-1β pathway, it diminished p38 phosphorylation, facilitating the restoration of Cx43 expression and mitigating cardiac electrical remodeling post-MI, consequently lowering cardiac susceptibility to VAs and the occurrence of HF (122). Furthermore, Phloretin also decreased the expression of fibrotic markers including collagen 1, collagen 3, TGF-β, and α-SMA post-MI by suppressing inflammatory responses orchestrated by NLRP3 inflammasome activation, consequently alleviating detrimental cardiac remodeling (122). The cardioprotective effects of scutellarin are mediated by its regulation of the Akt/mTORC1/NLRP3 signaling pathway (140). More precisely, scutellarin inhibits mTORC phosphorylation by upregulating Akt expression (140). This action subsequently diminishes the activation of the NLRP3 inflammasome, thus mitigating inflammatory injury and dysfunction in the heart induced by MI/R (140). Hydroxylsafflower yellow A (HSYA) was recognized for its ability to mitigate myocardial ischemia and hypoxic injury (141–143). In MI/R injury, HSYA suppressed the NLRP3 inflammasome by modulating the AMPK/mTOR signaling pathway, thereby reducing myocardial infarct size and decreasing cardiomyocyte apoptosis, ultimately improving heart function (143). In an H/R-induced H9c2 cell study, the AMPK inhibitor compound C nullified the suppressive impact of HSYA on NLRP3 inflammasome activation, as demonstrated by elevated levels of NLRP3, caspase-1, and IL-1β expression (142). This observation further corroborates that the inhibition of the AMPK/NLRP3 inflammasome signaling pathway is an important mechanism in the anti-MI/R injury effect of HSYA (142). In a study on oxygen-glucose deprivation/reoxygenation (OGD/R)-induced HUVECs, NLRP3 inflammasome-mediated pyroptosis was heightened (141). Treatment with HSYA mitigated pyroptosis by inhibiting the NLRP3/caspase-1/GSDMD pathway, thereby mitigating inflammatory damage to HUVECs resulting from OGD/R (141). Formononetin can alleviate MI/R injury (144). It restricts the activation of the NLRP3 inflammasome by diminishing the release of ROS, suppressing the expression of TXNIP, and attenuating the interaction between TXNIP and NLRP3, thereby decreasing the secretion of proinflammatory factors and cardiomyocyte apoptosis (144). Luteolin similarly demonstrated the potential to alleviate MI/R injury, and this protective attribute was associated with its suppression of the TLR4/NF-κB/NLRP3 inflammasome pathway (145). Luteolin downregulates the expression of TLR4, MyD88, and NF-κB in a dose-dependent manner to inhibit NLRP3 inflammasome activation, consequently diminishing myocardial infarct size and enhancing left ventricular function (145). Intriguingly, another study identified the SIRT1/NLRP3/NF-κB signaling pathway as the primary regulatory mechanism by which luteolin alleviates MI/R damage (146). These findings suggest that luteolin may exert cardioprotective effects by inhibiting the NLRP3 inflammasome through multiple molecular signaling pathways. Biochanin A alleviates the cardiac inflammatory response and reduces the infarcted myocardial area resulting from MI/R (147). Its cardioprotective effect was intricately linked to its inhibition of the TLR4/NF-κB/NLRP3 signaling pathway (147). By inhibiting NLRP3 inflammasome activation, irisin effectively restrained the expression of GSDMD-N and IL-1β, thereby mitigating the detrimental effects of pressure overload on the heart such as myocardial inflammation, fibrosis, and hypertrophy (148). By inhibiting the STING/NLRP3 signaling pathway, amentoflavone mitigates cardiomyocyte pyroptosis and cardiac inflammation, consequently ameliorating DOX-induced heart damage and functional impairment (149). Calycosin also shows promise for the treatment of myocardial toxicity (150). Mechanistically, it inhibited NLRP3 inflammasome activation by upregulating SIRT1 expression, thereby reducing cardiac inflammatory infiltration, myocardial fibrosis, and cardiomyocyte apoptosis, ultimately mitigating DOX-induced cardiac injury (150).
4.2 Terpenoids and their glycosidesAMI triggered intense inflammatory responses and oxidative stress (OS) (151). Gentiopicroside mitigates cardiac inflammatory responses, OS, and cardiomyocyte apoptosis induced by AMI by regulating the Nrf2/NLRP3 signaling pathway, thereby safeguarding cardiac function (151). In the pathological progression of chronic HF, Celastrol improves cardiac electrophysiological stability, upregulates Cx43 and ion channel expression, and reduces myocardial fibrosis and inflammatory responses by inhibiting the NLRP3/caspase-1/IL-1β signaling pathway, ultimately reducing susceptibility to ventricular fibrillation (16). Following MI, a notable increase was observed in macrophage and neutrophil infiltration of myocardial tissues alongside a significant upregulation in the expression of pro-fibrotic proteins such as collagen I, collagen III, and α-SMA (152). Celastrol mitigates these pathological alterations by inhibiting the NLRP3 inflammasome (152). Muscone exhibits a promising therapeutic potential against MI (123). It diminishes ventricular inflammation and fibrosis, while decreasing vulnerability to VAs via the upregulation of Cx43 expression in the infarct border zone (123). These effects were associated with its inhibitory impact on the NLRP3/IL-1β/p38 MAPK pathway (123). Furthermore, Muscone mitigated the macrophage-driven cardiac inflammatory response by suppressing NF-κB expression and NLRP3 inflammasome activation in myocardial macrophages, leading to enhanced cardiac function and increased survival rates in mice post-MI (153). Oridonin can alleviate cardiac remodeling post-MI (154). By inhibiting the NLRP3 inflammasome, it reduced the expression of fibrosis markers, including collagen-I, collagen-III, collagen-IV, and α-SMA, thereby alleviating myocardial fibrosis and cardiac dysfunction following MI (154). Moreover, pretreatment with oridonin suppressed the overactivation of OS and NLRP3 inflammasome, consequently mitigating cardiac pathological alterations induced by ischemia reperfusion, including the alleviation of myocardial inflammatory damage and reduction of infarct size (155). Sweroside inhibits the ROS-mediated NF-κB/NLRP3 inflammasome pathway in cardiomyocytes by directly binding to CaMKIIδ, alleviating myocardial inflammation and adverse cardiac remodeling, thereby improving HF induced by pressure overload (156). Sweroside also exerts protective effects on ischemia reperfusion myocardium (157). Its intervention alleviates myocardial inflammatory damage and reduces the size of the infarcted area, helping to alleviate cardiac dysfunction caused by MI/R (157). This effect is primarily due to the inhibition of NLRP3 inflammasome-mediated pyroptosis (157). Geniposide has therapeutic potential for alleviating MI/R injury (158). It inhibits the ROS/TXNIP/NLRP3 inflammasome pathway by activating the AMPK signaling pathway (158). This process efficiently suppresses cardiac inflammation, enhances myocardial energy metabolism, and ultimately reduces the damage inflicted on the myocardium by ischemia reperfusion (158). The glucagon-like peptide-1 receptor (GLP-1R)/NLRP3 pathway plays a pivotal role in mediating the cardioprotective effects of loganins (159). MI/R induces a notable decline in GLP-1R expression within the myocardial tissue, which promotes the formation of the NLRP3 inflammasome and pyroptosis, exacerbating myocardial damage and cardiomyocyte apoptosis (159). Conversely, treatment with loganin alleviates these pathological changes (159). Artemisinin pretreatment mitigates MI/R-induced myocardial inflammation, cardiomyocyte apoptosis, and myocardial fibrosis primarily by inhibiting the NLRP3 inflammasome (160). Betulin attenuated the cardiac inflammatory response, decreased myocardial infarct size, and enhanced cardiac electrical signaling by modulating the SIRT1/NLRP3/NF-κB signaling pathway. This action ultimately helps mitigate the cardiac pathological damage induced
留言 (0)