
記住我
Cutaneous squamous cell carcinoma (cSCC) is a malignant tumor originating from epidermal keratinocytes. It is one of the most commonly diagnosed non-melanoma skin cancers (NMSC), with its incidence gradually increasing worldwide in recent years, accounting for approximately 20% to 50% of all skin tumors (1, 2). In the United States, there are approximately 1.1 million new cases of cSCC annually, with an overall increase in incidence of 263% between 1976-1984 and 2000-2010 (3, 4). In western China, cSCC has the highest incidence rate among all skin cancers, at 29.4%, and this rate continues to rise annually by 2.6% (5). In European countries, it is estimated that by 2030, the incidence of cSCC will double (6). With a large number of cases, cSCC presents a significant burden on public health, with 3% to 7% of patients experiencing metastasis, leading to notable morbidity and mortality, thus imposing substantial societal public health burdens (3, 7, 8).
Actinic keratosis (AK) is a precancerous skin lesion caused by long-term chronic exposure to ultraviolet radiation (UVR), predominantly affecting sun-exposed areas such as the head and face, and commonly occurring in the elderly. AK carries a high risk of developing into cSCC and can present with multiple clinical and subclinical lesions simultaneously. The progression rate and probability of AK to cSCC vary among individuals, with an estimated annual progression rate of 0% to 0.075% per lesion (9). Among elderly patients with multiple AK lesions, the annual rate of progression to invasive cSCC is 0.6%, increasing to 2.57% four years after diagnosis (10).
The increasing incidence and mortality rates of cSCC have significant implications for public health. Understanding the malignant transformation from AK to cSCC is crucial for the prevention, early detection, and effective management strategies of cSCC. Additionally, a deeper understanding of the molecular mechanisms underlying the transition from AK to cSCC may facilitate the development of novel targets for the intervention of AK or cSCC.
2 Etiology, prevention, and early diagnosis of AKThe etiological factors of AK include environmental and individual factors. Environmental factors primarily refer to exposure to UVR, with the incidence of AK being positively correlated with cumulative UVR exposure (11, 12). Populations exposed to more sunlight (e.g., Australia and the United Kingdom) have a higher incidence of AK (13). UVR can induce cellular genetic mutations, chronic skin inflammation, and immune suppression, ultimately leading to abnormal proliferation of keratinocytes. The more sunlight the skin absorbs, the higher risk of AK (14). Five independent risk factors for AK are age (>60 years), gender (male), skin type, history of skin tumors, and outdoor work history (14). Individuals with fair skin (Fitzpatrick types I and II) are more prone to AK, while the incidence of AK is similar in Fitzpatrick types III and IV skin (12, 15). These conditions are influenced by various exposome factors such as air pollution, nutrition, and psychological stress, all of which can exacerbate skin carcinogenesis and aging (16). Additionally, immunosuppressed populations, such as 32.5% solid organ transplant recipients (SOTRs) (17) or patients taking long-term cytotoxic drugs, are at higher risk of AK (18–21).
Prevention of AK is strongly recommended, with evidence supporting using sunscreen and protective clothing to block initial and recurrent UVR radiation (22, 23). Randomized controlled trial (RCT) has shown that the use of sunscreen can reduce the incidence and progression of additional AK lesions (24). In addition to the significant role of sunscreen in AK prevention, oral medications also have a preventive effect on AK, such as oral nicotinamide (25–27), and acitretin (28, 29). However, a recent double-blind, randomized, placebo-controlled trial of oral nicotinamide for the prevention of AK in kidney transplant recipients showed that nicotinamide did not reduce the progression of AK in this population (30).
The diagnosis of AK emphasizes early detection and prediction of the risk of AK progression to cSCC. Non-invasive and highly efficient diagnostic techniques such as dermatoscopy, reflectance confocal microscopy (RCM), optical coherence tomography (OCT) (31), aminolevulinic acid (1%) (32), and high-frequency ultrasound of the skin (33) are employed for the diagnosis of AK. Dermatoscopy has good sensitivity and specificity for diagnosing AK, with a sensitivity of 95.6% and specificity of 95.0% for features such as the red pseudonetwork pattern combined with follicular pore enlargement (34). Dermatoscopic features of AK correlate with histological manifestations such as acanthosis, hyperkeratosis, and vascular proliferation. The “strawberry pattern” characteristic of non-pigmented AK can be detected by dermatoscopy and differentiated from early cSCC (35, 36). The most specific indications for diagnosing AK with RCM are the observation of disordered arrangements of keratinocyte layers and cellular atypia, with RCM having a unique diagnostic value for pigmented AK (12, 34). Histopathological examination remains the gold standard for diagnosing AK, especially when clinical diagnosis is uncertain or when progression to invasive cSCC is suspected. The main indications for pathological examination are uncertain clinical diagnosis, lesions > 1 cm in diameter, bleeding, ulceration, or induration, and rapid growth of skin lesions; secondary indications include lesions accompanied by severe itching, pain, and obvious hyperkeratosis (37). Skin lesions on certain special sites (such as the lips) are at high risk of invasion and require pathological examination. In addition, pathological subtyping helps guide the treatment and prognosis assessment of AK. Common pathological subtypes of AK include hypertrophic type, atrophic type, dyskeratotic type, mossy type, pigmented type, Bowenoid type, and actinic cheilitis type. Although AK histological subtypes have their characteristics, they mostly manifest as dysplastic keratinocytes arranged disorderly from mild basal layer to full-thickness epidermal structure disorder, appearing as a continuous spectrum structure of in situ squamous cell carcinoma, often accompanied by hyperkeratosis and dyskeratosis.
3 Etiology, prevention, and early diagnosis of cSCCcSCC shares similar risk factors with AK, including exposure to UVR, age, fair skin, and immunosuppression (38). UVR exposure stands out as the primary risk factor for cSCC. Prolonged sun exposure, cumulative exposure in specific areas, and incidents of sunburn are closely correlated with cSCC incidence, particularly among individuals with fair skin (38). The TP53 mutations induced by UVR represent the most common genetic alterations in cSCC (39). This malignancy is more prevalent among Caucasians, with an average onset age of approximately 60 years, and the risk increases with age, with a male-to-female ratio of about 3:1 (40). Immunocompromised individuals, such as SOTRs, face a significantly higher risk of developing cSCC, with SOTRs being 65 to 250 times more susceptible than healthy adults (41–43). Additionally, human papillomavirus (HPV) has been implicated in cSCC pathogenesis, particularly in cases affecting the perianal or genital regions (44). Environmental factors contributing to cSCC include arsenic exposure, polycyclic aromatic hydrocarbons, nitrosamines, and exposure to ionizing radiation (45, 46). Early signs of arsenic exposure include the occurrence of palmoplantar keratosis (45). Genodermatoses such as xeroderma pigmentosum, epidermodysplasia verruciformis, and epidermolysis bullosa dystrophica are also risk factors for cSCC development (47).
The “Interdisciplinary Guidelines for Invasive cSCC Based on European Consensus: Part 1 – Diagnosis and Prevention - 2023 Update” emphasizes primary and secondary prevention strategies for cSCC (48). Primary prevention involves avoiding sunbathing, using protective clothing, applying sunscreen, and seeking shade to minimize UVR exposure (48). Chemical prevention aims to reduce the incidence of new cSCC cases and includes agents such as nicotinamide, retinoids, 5-FU, and nonsteroidal anti-inflammatory drugs (NSAIDs). Nicotinamide can repair DNA damage and immunosuppression caused by intense UVR (49). However, a 12-month RCT involving SOTRs showed that oral nicotinamide did not reduce the incidence of cSCC (50). Several studies have demonstrated that oral retinoids, particularly acitretin and isotretinoin, can lower the incidence of new cSCC cases, especially among high-risk individuals (51, 52). An RCT on 5-FU showed that topical treatment reduced the expected surgical risk of cSCC by 75% (53). Population-based case-control studies suggest an association between NSAID use and a reduced risk of cSCC, with frequent NSAID use being a protective factor against cSCC development (54).
Digital cSCC lacks distinctive clinical features, leading to a potential misdiagnosis rate of up to 82% if solely based on clinical manifestations (55). Histopathological examination remains the gold standard for cSCC diagnosis, with well-differentiated histopathological subtypes exhibiting low metastatic potential, including keratoacanthoma and verrucous carcinoma, while aggressive subtypes associated with poor prognosis include desmoplastic cSCC, adenosquamous cSCC, and cSCC arising in association with scars (56, 57). Dermoscopy provides early diagnostic clues for cSCC and aids in the approximate assessment of its pathological staging, playing a crucial role in diagnosis, subsequent treatment, and management (58). cSCC lesions exhibit two vascular patterns under dermoscopy: dotted vessels and glomerular vessels. In situ pigmented cSCC may also present with small brown globules and homogeneous brown pigmentation under dermoscopy. Invasive cSCC often displays circular or serpentine vessels. RCM enables non-invasive diagnostic assessment of cSCC or AK skin lesions, with cSCC lesions showing a disordered arrangement of epidermal cells or atypical honeycomb patterns, along with circular vessels in the papillary dermis (59, 60).
4 Established progressed-associated genes from AK to cSCCDue to the fact that more than 70% of cSCC cases arise from AK (61), understanding the mechanisms underlying the progression from AK to cSCC is of paramount importance as it could aid in early therapeutic interventions before malignant transformation occurs. Identifying specific genomic changes that drive the transition from normal skin (NS) to AK and subsequently to invasive cSCC is challenging, given the substantial burden of UVR-induced mutations characteristic of all stages of this progression (Figure 1). Molecular genetic studies have examined AK and cSCC for known cancer gene expressions, chromosomal instability, and mutation levels (Figure 2). These studies generally indicate that AK and cSCC share similar genetic alterations (Table 1).
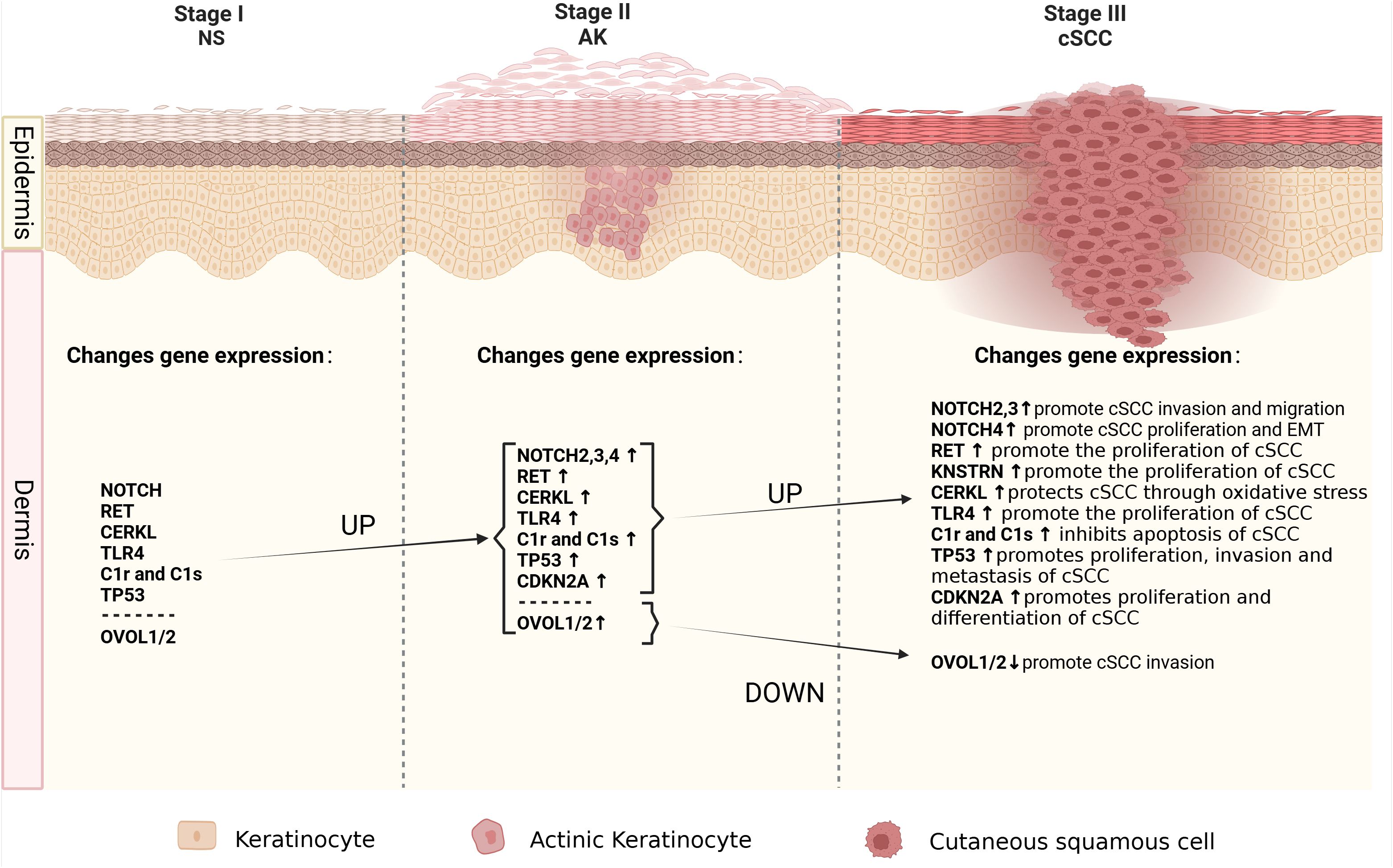
Figure 1. The molecular alterations associated with cancer progression from normal skin to AK to cSCC. Schematic depicting the spectrum of morphologies of NS-AK-cSCC lesions, with relevant changes in gene expression.
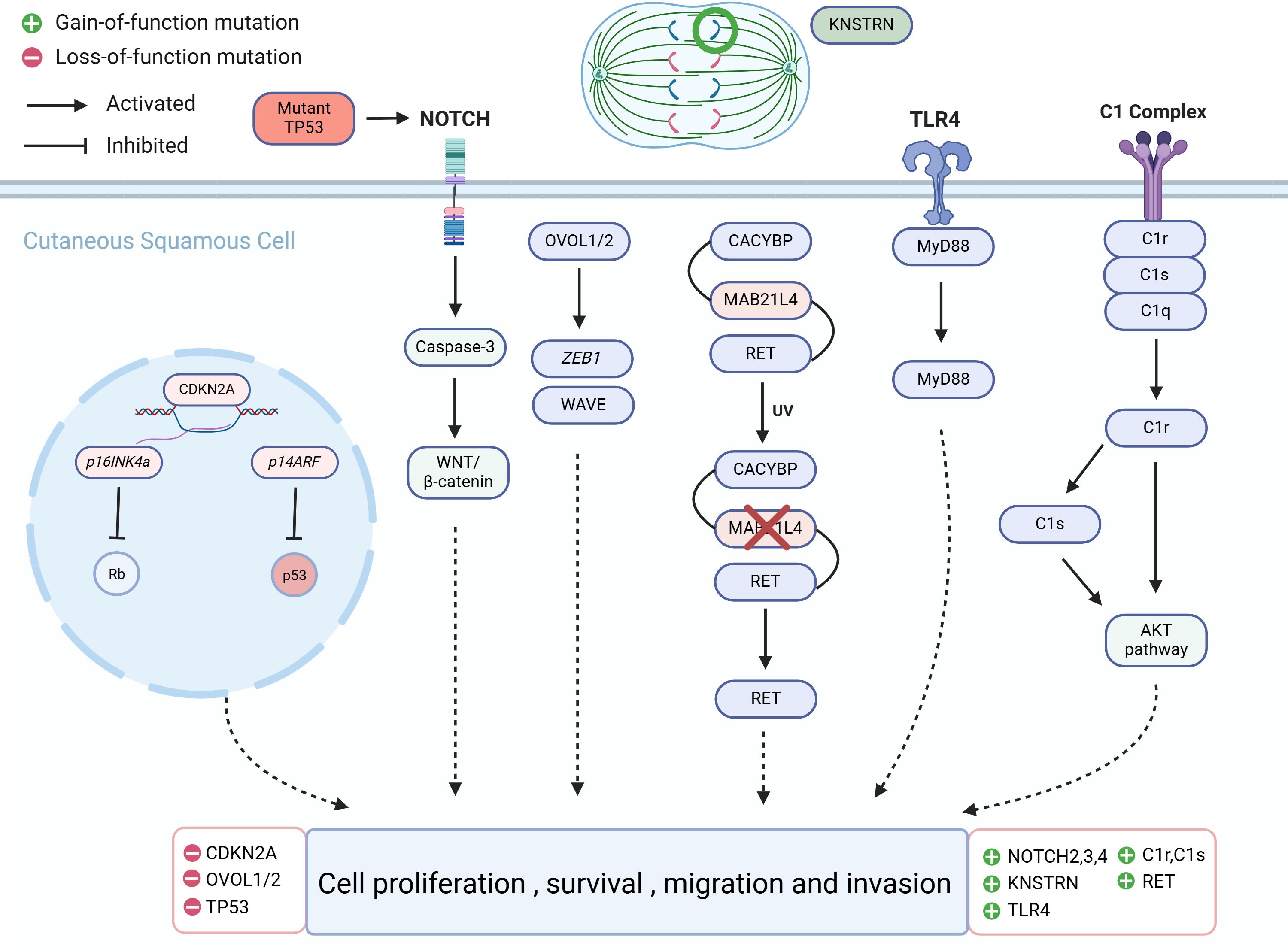
Figure 2. Molecular alterations that drive cutaneous squamous cell carcinoma (cSCC) proliferation.
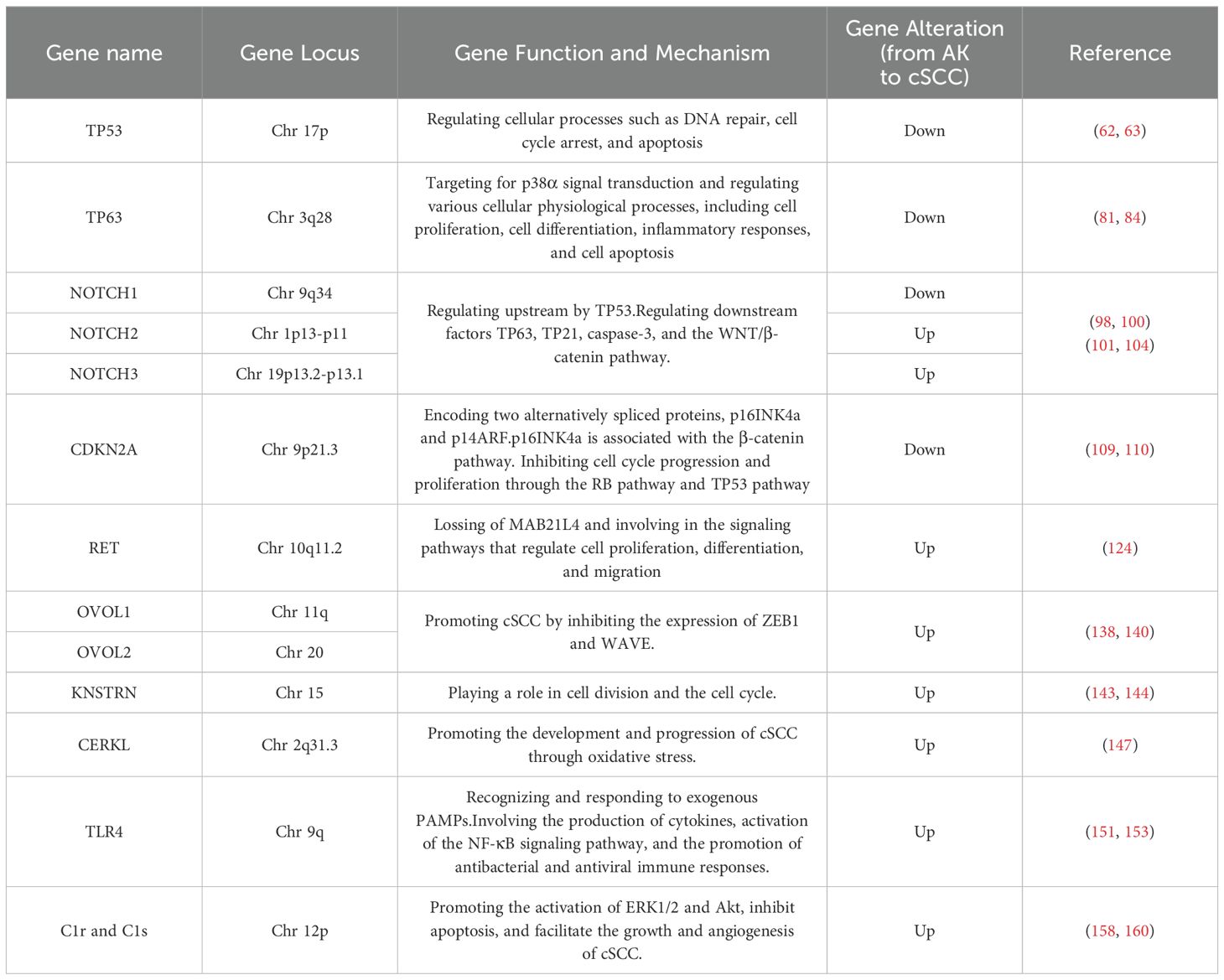
Table 1. The mutations and mechanisms in cSCC and AK genes across published studies.
4.1 TP53The tumor protein p53 (TP53) is situated on the short arm of chromosome 17 and encodes a 53-kilodalton protein involved in regulating cellular processes such as DNA repair, cell cycle arrest, and apoptosis (62, 63). TP53 is widely recognized as the most common mutated gene in cancer, being a pivotal tumor suppressor gene, whose abnormalities are associated with most malignancies (62). UVR-induced TP53 mutations are early events in cSCC development, leading to genomic instability, followed by mutations in other tumor suppressor genes and oncogenes (64). The mutation frequency of TP53 in primary cSCC ranges from 50% to 60%; in metastatic cSCC, TP53 mutations are present in nearly 95% of patients (65, 66). TP53 mutations are also present in normal skin exposed to UVR, AK, in situ, and invasive cSCC, indicating that TP53 mutations are not only early manifestations of UVR damage but also associated with cSCC progression (65, 66). Targeted deep sequencing of 160 cancer-related genes in 17 AK and cSCC patients revealed driver mutations in TP53 in 81% of cases, suggesting widespread TP53 mutations in AK and cSCC lesions (67). TP53 gene mutations have been found in a significant proportion of human skin malignancies, not only participate in the malignant transformation from AK to cSCC but also serving as biomarkers for predicting skin cancer (68). Skin cells exposed to ionizing radiation, such as solar UVR, incur DNA damage, leading to destabilization and inactivation of TP53 (69). TP53 gene mutations or alterations in the TP53 signaling pathway during the progression from AK to cSCC result in functional impairment of TP53, shifting its role from tumor suppression to oncogenesis, manifested as clonal proliferation of keratinocytes. The most typical pattern in AK is CC>TT base changes in abnormally proliferating keratinocytes, leading to further genetic instability and eventual cSCC development (65, 70). This clonal proliferation is considered a critical step in cSCC development (71). A deep sequencing study of skin clonal mutations demonstrated that TP53 R248W and G245D hotspot mutations are highly recurrent, potentially representing early stages of cSCC development (65).
Immunohistochemical results showed that the expression level of TP53 in cSCC was slightly lower than that of AK, and the expression level was significantly lower than that of NS (72, 73). Missense mutations occur in TP53 in cSCC, with common mutation hotspots such as TP53 R248W. Mutant TP53 gains oncogenic activity, serving as a potent early driver of cSCC progression. Additionally, missense mutations of TP53, such as p53 R172H, can lead to metastatic cSCC development (74). Besides missense mutations in TP53, some mutations result in nonsense mutations, thereby prematurely introducing a termination codon (75). Truncated isoforms produced by TP53 nonsense mutations include tumor-specific expressions characterized by increased metastasis, which may be a feature of tumor metastasis (76, 77). The functional mechanisms of TP53 mutant variants, including activation of cell proliferation, promotion of cell invasion and metastasis, and the presence and potential functions of different TP53 subtypes in cSCC development, are not yet fully understood.
Transcription factor (TF) TP63 is a p53 family member and plays a key role in the development of keratinocyte transformation induced by basal cell and ectodermal appendage carcinoma genes in stratified cSCC cell survival (78–82). p63 is a major regulator of epidermal development and maintenance and is also a key target for p38α signal transduction (81). The level of p63 protein detected in a large number of human cancer cell lines is negatively correlated with phosphorylated p38 signaling (81). P38α is an important signal-transducing protein kinase that belongs to the mitogen-activated protein kinase (MAPK) family (83). It plays a role in regulating various cellular physiological processes, including cell proliferation, cell differentiation, inflammatory responses, and cell apoptosis (79–81, 84). In cSCC biopsies, a significant reduction in p38 phosphorylation in the tumor parenchyma links AK with SCC progression and p38 inactivity (85). p38α plays a role in the transition from AK to cSCC, and p38α kinase is involved in the suppression of cSCC by regulating p63 and stem cell frequency (85).
TP53, as a pivotal gene, holds significant clinical relevance and applications in AK and cSCC. TP53 mutations are found in up to 100% of AK cases and 90% of cSCC cases, indicating its central role in the pathogenesis of these skin lesions (86). TP53 mutations are an early event that leads to the inactivation of its tumor suppressor function, facilitating the accumulation of additional oncogenic mutations and progression from AK to invasive cSCC (65). These mutations often result in a loss of normal p53 function and can lead to more aggressive tumor phenotypes (87). TP53 serves as a diagnostic and prognostic biomarker, the presence of TP53 mutations can serve as a diagnostic marker for AK and cSCC, as well as a prognostic indicator of disease progression and potential resistance to therapy (88). Reduced p53 staining in AK is associated with a higher likelihood of progression to cSCC, particularly in common and hypertrophic types of AK (86). Additionally, strategies to target TP53 mutations include the development of small molecules that reactivate mutant p53 or prevent its degradation, thereby restoring its tumor suppressor function (89). These approaches are promising in enhancing the efficacy of existing treatments and overcoming resistance in TP53-mutated tumors (90). However, not all TP53 mutations have the same biological impact. The loss of E-cadherin and increased expression of p53 are associated with the progression of AK to cSCC (91). The loss of E-cadherin and increased p53 are associated with AK progression, but it is important to consider the broader context of these molecular changes (91). While TP53 mutations are a hallmark of AK and cSCC, their role extends beyond tumor suppression to influencing therapeutic outcomes. The development of targeted therapies that address the specific mutations and pathways involved in TP53 dysfunction holds promise for improving patient outcomes. However, the complexity and variability of TP53 mutations necessitate a nuanced approach to treatment and research.
4.2 NOTCHThe NOTCH signaling pathway serves as a convergence point for numerous essential signaling pathways and is one of the most critical pathways determining cell fate. Signal transduction between adjacent cells via NOTCH receptors regulates cell differentiation, proliferation, and apoptosis, playing a widespread role in the occurrence and development of malignant tumors. Loss-of-function mutations in NOTCH1, NOTCH2, and NOTCH3 are commonly found in cSCC and sun-exposed skin, including AK, which does not exhibit microscopically evident dysplasia (66). NOTCH1, NOTCH2, and NOTCH3 genes are located on chromosomes 9q34, 1p13-p11, 19p13.2-p13.1, respectively (92). NOTCH signaling maintains the homeostasis of keratinocytes, and in AK, NOTCH1 mutations can occur in extracellular or transmembrane domains, as well as in intracellular domains. These mutations can disrupt the cellular microenvironment signaling both inside and outside the cell, leading to a predisposition to cancer (93–97). A genomic profiling study of AK revealed that NOTCH1 and NOTCH2 are significantly mutated genes, with mutation levels positively correlated with UVR intensity (98, 99). This study also found that NOTCH1 mRNA is significantly upregulated in AK compared to healthy skin, suggesting that NOTCH1 may have a carcinogenic role in AK, contrary to its presumed tumor-suppressive role in cSCC, where NOTCH1 is typically inactivated (98, 100). Further research is needed to elucidate the distinct functional roles of NOTCH1 in AK and cSCC progression.
Approximately 40% of cSCC patients exhibit inactivating mutations in NOTCH1, making it the second most common mutated gene after TP53 (101, 102). Studies indicate upregulation of NOTCH1 expression in cSCC, suggesting that NOTCH1 may function as both a tumor suppressor and a facilitator, exerting both tumor-suppressive and tumorigenic effects in cSCC (101). NOTCH2, NOTCH3, and NOTCH4 may act as oncogenes in cSCC, with studies showing high expression of NOTCH2 and NOTCH3 in cSCC tumor tissues. Upregulation of NOTCH2 expression may promote tumor cell migration and invasion, while high expression of NOTCH3 may be positively correlated with tumor cell proliferation and volume (101). NOTCH4 is upregulated in cSCC through the NOTCH4/HEY1 pathway, inducing tumor cell proliferation, resistance to cisplatin, and promoting epithelial-to-mesenchymal transition (EMT) (103). The expression of NOTCH genes is associated with upstream gene TP53 and downstream regulatory factors TP63, TP21, caspase3, and the WNT/β-catenin pathway, although the specific mechanisms remain unclear (66, 101).
The clinical application of targeting the NOTCH gene in AK and cSCC involves understanding its dual role as both an oncogene and a tumor suppressor, depending on the cellular context (104). This dual role complicates the therapeutic targeting of NOTCH, as its inhibition could potentially have opposing effects depending on the specific cancer type and stage (105). Gamma-secretase inhibitors (GSIs) are a class of drugs that inhibit the activation of NOTCH receptors by preventing the cleavage of the NOTCH intracellular domain (106). Furthermore, given the complexity of NOTCH signaling, combination therapies that target multiple pathways are being investigated. For instance, combining NOTCH inhibitors with other signaling pathway inhibitors, such as those targeting the PI3K/Akt or VEGF pathways, may enhance therapeutic efficacy (105). These inhibitors are being explored in clinical trials for their potential to suppress tumor growth in cancers with aberrant NOTCH signaling (107). Nevertheless, inhibiting NOTCH signaling can have unintended consequences due to its role in normal cellular processes (104). Therefore, careful consideration of the potential side effects and the development of selective inhibitors are crucial for successful clinical application.
4.3 CDKN2AThe CDKN2A gene is situated on chromosome 9p21.3, a region known for its involvement in tumorigenesis across different cancer types (108). It encodes two alternatively spliced proteins, p16INK4a and p14ARF, which inhibit cell cycle progression and proliferation through the RB pathway and TP53 pathway, respectively (98, 109, 110). When CDKN2A is highly expressed, it inhibits the proliferation of tumor cells. However, mutations or inactivation of CDKN2A can diminish its ability to suppress tumor cells, leading to rapid proliferation and differentiation (111). Exome sequencing studies of AK have identified CDKN2A as a non-candidate driver gene (98). Previous research also suggests that CDKN2A does not mutate in normal skin and may serve as a gatekeeper in the AK-cSCC transition (112). Genomic studies of somatic mutations and copy number alterations in AK and cSCC have found that CDKN2A driver mutations are more common in cSCC (30.8%) than in AK (11.1%), indicating fewer genetic mutations in CDKN2A in AK. However, CDKN2A is located on chromosome band 9p21, one of the most common genomic alteration regions in human cancers (113). Loss of heterozygosity at 9p21, leading to CDKN2A mutations or homozygous deletion, results in loss of function, which may contribute to the AK-to-cSCC transition (109, 114). Mutation analysis of CDKN2A loci in 26 AK specimens revealed new mutations, additional mutations, and missense mutations, corresponding to four different nucleotide changes, three in exon 1a of p16INK4a and one in exon 2 (115).
CDKN2A is strongly associated with the occurrence of skin malignancies. As a tumor suppressor gene, CDKN2A negatively regulates the cell cycle and promotes cellular senescence. Knockout mouse experiments suggest that p14ARF may play a role in the initiation of cSCC development, while p16INK4a may be involved in determining the differentiation status of cSCC (109, 116). Recurrent CDKN2A mutations have been found in skin malignancies, and inactivation of CDKN2A at the locus may be due to allelic loss, point mutations, and promoter hypermethylation. Inactivation of the CDKN2A locus leads to loss of control over the cell cycle and cell growth (117). Heterozygous loss or point mutations have been detected in 21% to 62% of cSCC cases, and promoter hypermethylation has been found in 35% to 78% of cases. Recently, immunohistochemical analysis of cSCC precursor lesions revealed a linear correlation between the immunostaining of p16INK4a and the membranous immunostaining of β-catenin in AK, indicating the importance of p16INK4a in the progression of cSCC precursor lesions (118).
CDKN2A mutation and expression levels serve as valuable prognostic markers and potential therapeutic targets. CDKN2A mutations are significantly associated with disease-specific death in metastatic cSCC, indicating its potential as a prognostic biomarker. Patients with CDKN2A mutations exhibit shorter disease-specific survival compared to those without such mutations (119). In cSCC, CDKN2A mutations are early genetic events, often occurring alongside TP53 mutations (119). These mutations are present in both primary tumors and metastases, suggesting their role in the early stages of tumorigenesis. The presence of CDKN2A mutations correlates with adverse clinical outcomes, such as increased tumor size and invasion depth, which are significant predictors of poor survival in cSCC patients (119). In the context of CDKN2A-associated therapeutic approaches, CDKN2A expression is linked to immune cell infiltration and immune regulation pathways, which could influence the tumor microenvironment and response to immunotherapy (120). The gene’s role in immune regulation and its association with immune checkpoints like PD-1 suggest that CDKN2A could guide immunotherapy strategies, potentially improving responses in cancers where it is highly expressed (121). The gene’s ability to influence tumor progression and response to therapies highlights its importance as a therapeutic target and biomarker. However, further research is needed to explore its specific applications in AK and cSCC, considering the unique pathophysiology of these skin conditions.
4.4 RETRET (Rearranged during transfection) is located on chromosome 10q11.2 and is involved in the signaling pathways that regulate cell proliferation, differentiation, and migration (122). Through different splicing mechanisms, RET can undergo 3′-end cleavage to form various receptor tyrosine kinases, including RET9, RET43, and RET51, which constitute the single-pass transmembrane tyrosine kinase receptor protein RET family. These isoforms exhibit tissue-specific expression (123). The exact role of RET in AK and cSCC remains unclear. It is activated by binding with soluble glial cell-derived neurotrophic factor family ligand proteins, leading to the production of RET proteins with abnormal activity. These aberrant signals may influence processes such as cell growth, survival, invasion, and metastasis. In AK, RET is significantly expressed and can drive cSCC, and the activation mechanism of RET is associated with the loss of MAB21L4 (124). MAB21L4, also known as Male Abnormal 21-like 4 or C2orf54, plays a role in differentiating human progenitor epidermal keratinocytes (125). MAB21L4 is considered a bridging protein between CACYBP and RET. In the presence of MAB21L4, the binding affinity between CACYBP and RET increases by 44 times. CACYBP can bind to the E3 ubiquitin ligase SIAH1 and participate in various cellular processes, including cell differentiation, proliferation, cytoskeletal dynamics, and tumorigenesis (126). High expression of RET in AK is caused by the loss of MAB21L4, which is an early event in AK. This loss disrupts the proximity of RET to the CACYBP-SIAH1 complex, preventing RET ubiquitination (124).
While the primary focus of RET inhibitors has been on lung and thyroid cancers, their application in skin cancers like AK and cSCC is an area of interest. The application of RET inhibitors in treating these cancers has evolved significantly, with a focus on developing selective inhibitors to improve efficacy and reduce side effects. This approach is particularly relevant in the context of AK and cSCC, where targeted therapies are increasingly being explored (127). The development of selective RET inhibitors like selpercatinib and pralsetinib has marked a significant advancement in targeting RET-driven cancers (128). These inhibitors have shown high efficacy and favorable toxicity profiles compared to earlier multikinase inhibitors (MKIs), which had off-target effects and limited clinical activity (129). Selpercatinib and pralsetinib have been approved by the FDA for treating RET-altered cancers, demonstrating high response rates in clinical trials (130). These inhibitors are effective against RET gatekeeper mutations, which are common resistance mechanisms in cancer therapy (131). The exploration of RET inhibitors in skin cancers is still in its early stages. However, the success of these inhibitors in other cancer types suggests potential applicability, especially in cases where RET alterations are identified (132).
4.5 OVOL1/2OVOL1 and OVOL2 (OVOL1/2) belong to the OVO gene family and are located on chromosomes 11 and 20, encoding a class of DNA-binding proteins that typically function as transcription factors regulating gene expression, acting as both transcriptional activators and repressors (84, 133–136). Three members of the OVO gene family have been identified: OVOL1, OVOL2, and OVOL3 (137). Among them, (OVOL1/2) have been studied most extensively. OVOL1/2 participate in suppressing EMT in tumor cells (133). They can inhibit the expression of Zinc finger E-box binding homeobox 1 (ZEB1) in tumor cells (138). OVOL1 levels are significantly elevated in non-metastatic oral squamous cell carcinoma tissue and negatively correlated with ZEB1 levels. OVOL2 is significantly downregulated in metastatic nasopharyngeal carcinoma cells, which exhibit strong invasive capabilities. Knockdown of ZEB1 can reduce the invasion capability of these cells (139). In 30 AK and 30 cSCC cells, knocking down OVOL1/2 increased the mRNA expression of ZEB1 and WAVE proteins. Specifically, knocking down OVOL1 significantly increased the expression of ZEB1 and WAVE proteins, while knocking down OVOL2 significantly increased the expression of ZEB1. Loss of OVOL2 significantly enhanced the invasion capability of cSCC cells (140). Immunohistochemical analysis of AK and cSCC cells showed that the expression of OVOL1 and OVOL2 was upregulated in AK but significantly downregulated in cSCC. Conversely, ZEB1 and WAVE proteins were upregulated in cSCC, indicating that the downregulation of OVOL1/2 and the upregulation of ZEB1 and WAVE proteins may be associated with the progression of AK to cSCC (140).
The application of OVOL1 and OVOL2 genes in the treatment of AK and cSCC is a promising area of research due to their roles in modulating EMT. This makes them attractive targets for therapeutic strategies aimed at halting or reversing the progression of these skin lesions. Targeting the OVOL2/ZEB1 axis could be a viable therapeutic approach to prevent the development of cSCC from AK. By inhibiting ZEB1, OVOL2 can potentially reduce the invasiveness of cSCC cells, making it a promising target for drug development (140). While the potential of OVOL1 and OVOL2 in treating AK and cSCC is promising, it is important to consider the complexity of cancer progression and the multitude of genetic and environmental factors involved. Therefore, while targeting OVOL1 and OVOL2 could be beneficial, a comprehensive approach that considers these other factors may be necessary for effective treatment.
4.6 KNSTRNKNSTRN is a protein that binds to Astrin on the kinetochore in the late stage of cell mitosis and is closely related to cell division (141). The KNSTRN gene is located on chromosome 15 at the 15q15.1 locus. It encodes a protein that is essential for proper chromosome alignment and segregation during cell division, particularly mitosis (142). KNSTRN is believed to play a role in cell division and the cell cycle process. Recurrent mutations were detected in 19% of AK and cSCC, mainly concentrated in KNSTRN mutations (143). Exome sequencing of 112 cSCC genes showed that the mutation sites were located at the N-terminus. 21 out of 112 (18.7%) patients had somatic mutations in KNSTRN, with over half of the mutations occurring at codon 24, resulting in a change from serine to phenylalanine due to a C-T transition (143). Detection of the frequency of KNSTRN gene mutations in healthy skin, photo-damaged skin, AK, and invasive cSCC with different histological grades showed that recurrent somatic mutations in KNSTRN p.Ala40Glu were associated with basal proliferative AK in invasive squamous cell carcinoma (144). These studies also suggest that KNSTRN may play a driving role in the occurrence and development of cSCC and could be a useful predictor for cSCC development.
The KNSTRN gene has emerged as a significant player in the development and progression of AK and cSCC. Mutations in this gene, particularly the recurrent somatic mutations, have been linked to the acceleration of cSCC development and are associated with different histological classifications of AK. These findings suggest that targeting the KNSTRN gene could be a promising strategy for treating these skin conditions. Given the role of KNSTRN mutations in cSCC progression, therapies targeting these mutations could potentially halt or reverse the disease process. This could involve the development of small molecules or biologics that specifically inhibit the mutated forms of the KNSTRN protein (140). Techniques such as CRISPR-Cas9 could be employed to correct KNSTRN mutations in affected cells, potentially preventing the progression of AK to cSCC (140). While KNSTRN mutations are prevalent in cSCC, they are not exclusive to this cancer type, as they have also been found in other cutaneous tumors like malignant melanomas (140). This necessitates a careful approach to ensure that therapies targeting KNSTRN mutations do not adversely affect normal tissues.
4.7 CERKLThe equations should be inserted in editable format from the equation editor. Ceramide kinase-like (CERKL) is situated on chromosome 2q31.3, and it is an oxidative stress-related protein whose expression is typically limited to the retina, nervous tissue, kidneys, trachea, testes, and lungs (145, 146). Levels of CERKL in normal skin are relatively low, but CERKL mRNA and protein are highly expressed in AK and cSCC (147). Knocking down CERKL in cSCC cells reduces the content of cell sheath lipids and enhances the sensitivity of cSCC cells to oxidative stress, indicating that CERKL may protect cSCC cells through an oxidative stress protection mechanism (147). The mechanism of oxidative stress protection mediated by CERKL is currently unclear but it may involve CERKL-mediated regulation of cellular autophagy (148). The mechanisms of CERKL in AK and cSCC require further investigation.
Studies have identified various genes involved in cSCC progression, including CERKL, which could serve as biomarkers for early diagnosis and treatment strategies (149). CERKL is highly expressed in cSCC and AK compared to normal epidermis. It plays a role in maintaining cellular sphingolipids and protecting cells from oxidative stress, which are critical in the survival and proliferation of cancer cells (149). By enhancing resistance to oxidative stress, CERKL may contribute to the survival of cancer cells under hostile conditions, making it a potential target for therapies aimed at increasing cancer cell susceptibility to oxidative damage (149). While CERKL presents a promising target for cSCC and AK treatment, it is essential to consider the complex genetic and molecular landscape of these conditions.
4.8 TLR4Toll-like receptor 4 (TLR4) is located on chromosome 9q in humans, and it also is a receptor protein in the human immune system (150). It belongs to the Toll-like receptors (TLRs) family and plays a major role in regulating immune responses and inflammatory responses. TLR4 protein is mainly expressed on the surface of immune cells such as macrophages, dendritic cells, and certain epithelial cells. Its main function is to recognize and respond to pathogen-associated molecular patterns (PAMPs) from exogenous pathogens. TLR4 binds to these PAMPs and activates immune cells, triggering a series of immune responses, including the production of cytokines, activation of the NF-κB signaling pathway, and promotion of antibacterial and antiviral immune responses. TLR4 is expressed in human skin, AK, and cSCC (151). TLR4 signal transduction promotes cSCC development in a MyD88-dependent manner and is essential for inflammatory cell recruitment during carcinogenesis (152). TLR4 may play a tumor-promoting role in cSCC, and immunohistochemical detection shows that the expression of TLR4 in cSCC is significantly higher than that in AK tissue, and its expression in low-differentiated cSCC is significantly lower than that in moderately differentiated and highly differentiated cSCC (153).
The clinical application of the TLR4 gene in AK and cSCC involves understanding its role in tumor progression and immune response modulation. TLR4 antagonists, such as eritoran and resatorvid, have been identified as potential therapeutics. These agents can suppress UV-induced inflammatory signaling, which is crucial in preventing skin photodamage and photocarcinogenesis (154). On top of that, TLR agonists have been used in immunotherapy for skin cancers, including melanoma and basal cell carcinoma, by enhancing dendritic cell recruitment and T-cell responses (155). This approach could be adapted for cSCC, targeting TLR4 to modulate immune responses (156).
4.9 C1r, C1s, and factor DC1r and C1s are two important components of the complement system, which belong to the C1 complex. Both C1r and C1s genes are located on chromosome 12, specifically in the region 12p13 (157). The C1 complex is the initial enzyme complex of the complement system and participates in the regulation of immune responses and inflammatory reactions. C1r and C1s are both enzyme proteins that play a key role in the C1 complex. C1r plays the role of an initiator enzyme in the activation of the C1 complex. When C1r is activated, it catalyzes the activation of C1s (158). C1s, on the other hand, is responsible for further activating other components of the complement system, such as C4 and C2 (159). Compared with normal epidermal keratinocytes, the mRNA levels and production of C1r and C1s are significantly increased in cSCC cells (160). Reducing the expression of C1r and C1s in cSCC cells suppressed the activation of extracellular signal-regulated kinase 1/2 and Akt, promoting cSCC cell apoptosis and significantly inhibiting the growth and angiogenesis of human cSCC xenografts (158). Furthermore, C1r and C1s were highly expressed in aggressive disseminated cSCC and cSCC associated with pemphigus vulgaris, while they were expressed less in in situ cSCC, AK, and normal skin (160).
Given their role in promoting cSCC growth, C1r and C1s represent potential targets for clinical application therapeutic intervention. As markers of aggressive tumor behavior, C1r and C1s could help predict the likelihood of progression from AK to invasive cSCC, aiding in risk stratification and management decisions (160). Inhibitors or monoclonal antibodies against these proteins could be developed to suppress tumor growth and improve patient outcomes (160). The integration of C1r and C1s targeting into clinical practice will require further validation in larger cohorts and the development of specific inhibitors.
Complement factor D(FD) cleaves C3 to produce the active fragments C3a and C3b. C3a can induce inflammatory reactions, causing vasodilation and promoting the infiltration of inflammatory cells (161–163). In culture and in vivo, cSCC cells express FD significantly. The in vivo FD expression level in non-metastatic cSCC, metastatic cSCC, cSCC metastasis, and pemphigus vulgaris-related cSCC is significantly higher than that in normal skin, precancerous lesions, AK, and in situ cSCC. The strongest FD expression is limited to the invasive edge of the tumor, while the weak FD staining pattern in normal skin, AK, and cSCC in situ shows a diffuse and uniform distribution (164).
FD’s role in the alternative complement pathway makes it a promising target for therapeutic intervention. The small-molecule FD inhibitor Danicopan has been proposed as a specific drug candidate for advanced cSCC therapy (164). Inhibition of FD activity has been shown to attenuate cSCC cell proliferation, suggesting that targeting FD could be an effective strategy for managing cSCC progression (164). While the role of FD is promising in the treatment of cSCC, further research is needed to fully elucidate the complex interactions between these factors and to validate FD’s clinical utility in larger, diverse patient populations.
5 Nomenclature established progressed-associated non-coding RNAs from AK to cSCCRNA, as an integral part of gene regulation, has garnered widespread attention, with non-coding RNA (ncRNA) emerging as a focal point of research in recent years. Variants such as long non-coding RNA (lncRNA) and microRNA (miRNA) have been particularly emphasized. The precise mechanisms and specific targets of ncRNA are still actively under investigation, as they serve as potential regulatory molecules with significant roles in skin cancer. Further research endeavors will contribute to a better understanding of the exact functions of RNA molecules in the progression from AK to cSCC and their potential clinical applications.
5.1 Long non-coding RNALong non-coding RNA (lncRNA) refers to a group of endogenous RNAs with lengths exceeding 200 nucleotides, lacking complete open reading frames, and thus not encoding proteins (165). However, lncRNA can interact with DNA, RNA, and proteins through various mechanisms, participating in related biological processes such as serving as protein scaffolds, directly interacting with proteins and RNA (166), participating in chromatin modification, regulating mRNA, transcription factors, and epigenetics, and acting as competing endogenous RNA for microRNA (167). A study employing RNA sequencing of two cases of AK and three cases of normal skin tissue identified lncRNA uc011fnr.2 negatively regulating SCIMP and TLR4, and being involved in activating the JAK-STAT3 signaling pathway in AK (168). Additionally, the dysregulation of lncRNAs in premalignant lesions like actinic keratosis (AK) suggests their involvement in the progression to invasive cSCC (168).
5.2 miRNAmiRNAs are short non-coding RNAs that regulate protein-coding gene expression at the post-transcriptional level. miRNAs play important roles in the initiation and progression of tumorigenesis by regulating the expression of tumor suppressor genes and oncogenes (169–171). The most significant changes in miRNA expression occur during the transition from “early” stages of normal epidermis, AK, and early photodamaged epidermis to the “late” stages of epidermal carcinogenesis, late-stage AK, and cSCC (172). By analyzing the differential expression of genes in 10 AK and 30 cSCC tissue samples, using microarray dataset GSE45216 obtained from the gene expression database, they identified 16 miRNAs and found that three co-expressed network modules EIF4EBP1, SNX17, PRPF4, NXT1, and UBA5 may be pathogenic genes contributing to the development of AK into cSCC (173). Recent studies have also demonstrated that miRNA regulation plays a critical role in the progression of epidermal keratinocytic neoplasms (174, 175).
miR-204 primarily functions as a tumor suppressor gene. It has been shown that DNA methylation in the upstream promoter region of TRPM3, a gene upstream of miR-204, is one of the mechanisms contributing to the silencing of miR-204 in cSCC (176). Comparison of miR-204 expression between cSCC, AK, and healthy skin tissues, revealed significantly downregulated miR-204 in cSCC. Overexpression of miR-204 inhibits the activation and translocation of transcription activator 3, thereby suppressing the development of cSCC. Protein tyrosine phosphatase non-receptor type 11 (PTPN11) has been identified as a direct target of miR-204 (176). miR-23b is downregulated in both AK and cSCC, and its expression is regulated by the MAPK signaling pathway. miR-23b reduces the expression of FGF2 at both mRNA and protein levels, impairing the ability of cSCC cells to induce angiogenesis. Overexpression of miR-23b inhibits colony and sphere formation in cSCC cells, while CRISPR/Cas-9-mediated deletion of miR-23b leads to increased colony and tumor sphere formation in vitro (177). Experimental studies have confirmed the overexpression of Ras-related protein RRAS2 in cSCC, and the interference of RRAS2 expression impairs angiogenesis, colony formation, and tumor sphere formation. Importantly, RRAS2 is a direct target of miR-23b in cSCC (177). miR-181a acts as a transcriptional driver factor in the progression from AK to cSCC. miR-181a promotes the formation of various proteins by targeting the TGFβ receptor 3 (TGFβ R3) in the TGFβ signaling pathway. In cSCC, the expression level of miR-181a is inversely correlated with the expression level of TGFβ R3. Overexpression of miR-181a or knockdown of TGFβ R3 inhibits apoptosis and enhances cell survival. Moreover, miR-181a enhances cell migration and invasion and upregulates epithelial-mesenchymal transition markers. miR-181a regulates these processes by directly interacting with TGFβ R3. Experimental results have demonstrated that miR-181a inhibits tumor growth, and reversing these effects can be achieved by increasing or decreasing TGFβ R3 expression (178). Additionally, a study has shown the involvement of miRNA-31 and MMP-1 in differentiation pathways associated with increased EMT and angiogenesis, which are important mechanisms in cSCC invasion. EMT is enhanced in AK (179). These findings hold significant implications for future research on targeted therapies.
6 Application of clinical drugs in AK and cSCCThe treatment of AK and cSCC involves a variety of clinical drugs and therapeutic approaches. These conditions, which are closely related, often require a combination of topical, systemic, and procedural treatments to manage effectively. The choice of treatment depends on factors such as the severity of the lesions, patient characteristics, and the potential for malignant transformation. Below is an overview of the current clinical drugs used in the management of AK and cSCC (Figure 3).
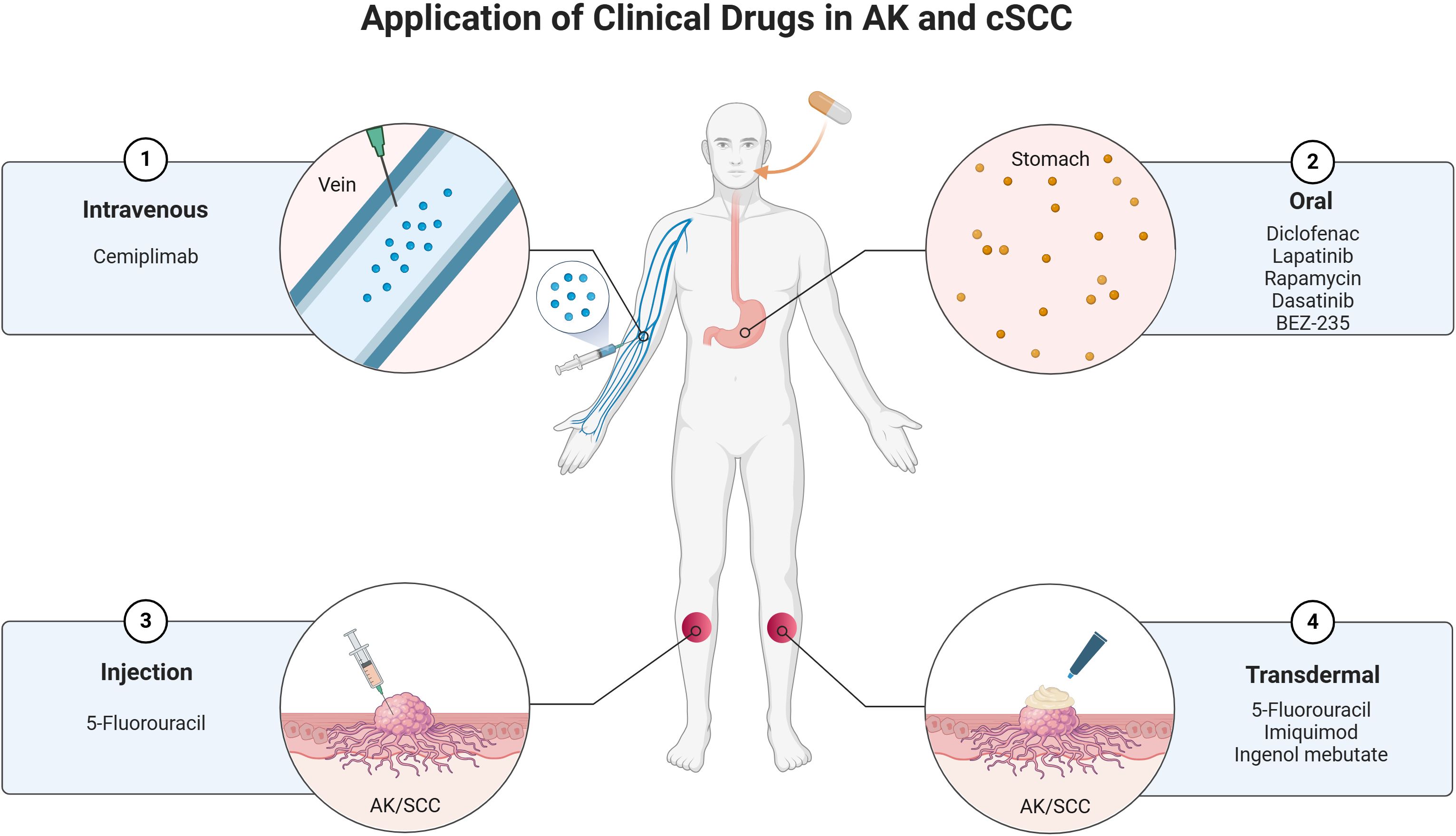
Figure 3. Current clinical drugs used in the management of AK and cSCC.
6.1 Topical treatments6.1.1 5-fluorouracilThis is a widely used topical chemotherapeutic agent for AK and cSCC (180). A novel 4% formulation of 5-FU cream has shown high efficacy in treating multiple AKs, with significant reductions in the Actinic Keratosis Area and Severity Index (AKASI) scores observed at 6 and 12 weeks post-treatment (181). It works by inhibiting DNA synthesis in rapidly dividing cells, leading to the eradication of AK and cSCC lesions (182). Combining 5-FU with photodynamic therapy (PDT) has been shown to enhance lesion clearance rates (183). Pretreatment with 5-FU increases protoporphyrin IX accumulation, improving the efficacy of PDT in AK treatment (183). Unlike its use in the treatment of AK, 5-FU has alternative approaches for managing cSCC. Intralesional 5-FU at lower concentrations (10.0 mg/mL and 16.7 mg/mL) has been effective in treating cSCC and keratoacanthomas, achieving a 96% lesion clearance rate with minimal adverse effects (184). This approach strategically reduces the cumulative dose of 5-FU, thereby mitigating potential toxicities without compromising therapeutic efficacy. Furthermore, a combinative treatment strategy involving debulking surgery followed by intralesional 5-FU has evinced robust tumor clearance rates, particularly in instances of well-differentiated SCCs (185). This integrated method has proven especially efficacious for managing tumors located on the trunk and extremities, offering a tailored and effective treatment modality for such cases. While 5-FU has proven effective in treating AK and cSCC, its application requires careful consideration. The potential for combination therapies, such as with PDT, offers promising avenues for enhanced treatment outcomes. However, the variability in response rates and the need for individualized treatment plans underscore the complexity of managing these conditions. Further research is needed to optimize 5-FU use across diverse patient groups and to explore its full potential in preventing progression to invasive cSCC.
6.1.2 ImiquimodImiquimod is widely used for AK treatment due to its ability to modulate the immune system, leading to the clearance of lesions. This immune response modifier is used in 5% cream form for AK treatment (186). It works by inducing cytokine production, which promotes apoptosis in abnormal keratinocytes (187). Studies have shown clearance rates ranging from 66% to 87% when used as monotherapy (188). However, combining imiquimod with other agents like 5-fluorouracil has been explored to enhance efficacy (188). This combination has shown higher clearance rates and shorter treatment durations, suggesting a synergistic effect (188). In the treatment of cSCC, imiquimod has been used off-label for treating cSCC, particularly in cases where surgery or radiation is not feasible (189). A notable case involved the successful treatment of a locally advanced cSCC of the eyelid with imiquimod 3.75%, resulting in complete tumor regression without significant side effects (189). While imiquimod has demonstrated efficacy in treating AK and cSCC, its application is not without challenges. The variability in patient response, potential for local skin reactions, and the need for further studies to confirm long-term outcomes and optimal treatment regimens are areas that require attention. Overall, imiquimod remains a valuable tool in dermatological oncology, with ongoing research likely to expand its utility and effectiveness.
6.1.3 DiclofenacDiclofenac, a non-steroidal anti-inflammatory drug (NSAID), has been explored for its application in treating AK and cSCC. Diclofenac’s mechanism involves the inhibition of cyclooxygenase-2 (COX-2), reducing prostaglandin E2 levels, which are implicated in tumorigenesis and inflammation associated with AK (190). And the drug also induces apoptosis and alters cell cycle profiles in cancerous cells, potentially through p53 induction and metabolic alterations, as observed in esophageal squamous cell carcinoma models (191). Diclofenac 3% gel, often combined with hyaluronic acid, is a topical treatment for AK. It has been shown to be effective, with a resolution of at least 50% of lesions in 78% of patients, and a high satisfaction rate of 85% (192). But in a comparative study, diclofenac was less effective than imiquimod in preventing the progression of AK to more severe forms or invasive SCC over a three-year period (192) While diclofenac offers a viable option for treating AK and potentially cSCC, its role is often complementary to other therapies. Its ability to modulate immune responses and cellular metabolism presents a unique advantage, but its efficacy in preventing progression to invasive SCC is less than some alternatives. Future research may focus on optimizing its use in combination therapies and exploring its full potential in cancer prevention and treatment.
6.1.4 Ingenol mebutateIngenol mebutate is FDA-approved for treating AK, a p
留言 (0)