
記住我


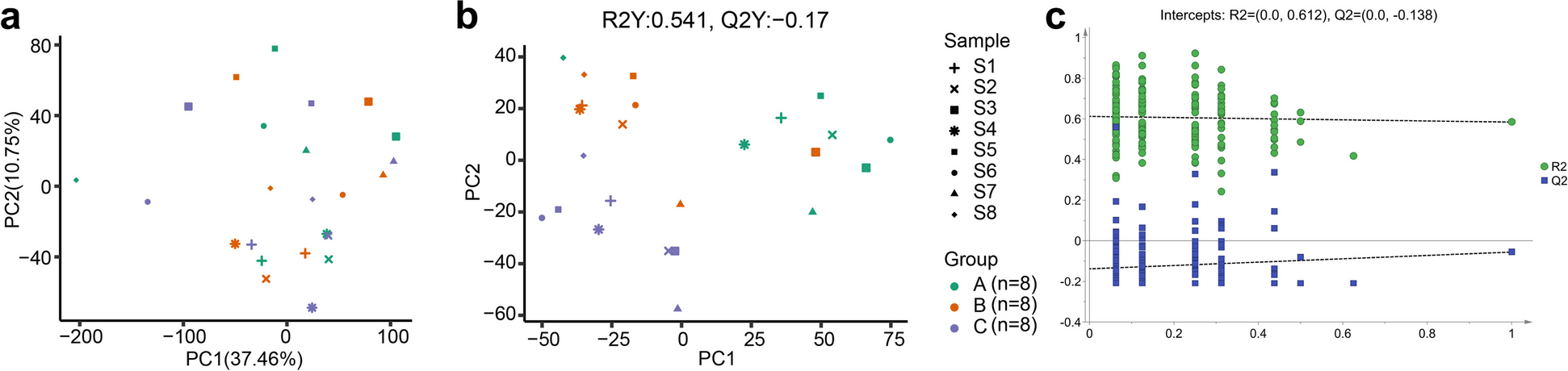



To determine systematic differences in the transcriptome landscape before and after entering into the simulated high-altitude environment, RNA-seq was performed on blood samples derived from 8 volunteers, including after adapting to an altitude equivalent to sea level for 1 h (baseline, group A, n = 8), after adapting to an altitude 3500 m for 1 h (group B, n = 8), and after adapting to an altitude 4500 m for 1 h (group C, n = 8). At first, PCA was carried out based on the gene expression landscape. As shown in the score plot, samples from three groups were indistinguishable, which was further confirmed by PERMANOVA analysis (sample: R2 = 0.448, P = 0.009; group: R2 = 0.057, P = 0.611) (Fig. 1a). To separate the groups, supervised PLS-DA was carried out by fitting these samples. The PLS-DA model organized the replicates into three distinct clusters corresponding to the A, B, and C groups, with few outlier samples (Fig. 1b). Although the model fitted relatively well to the data set (R2Y value 0.541), the predictive performance of the model was poor (Q2Y value − 0.17). Besides, the result of permutation test indicated that the model was overfitting (Fig. 1c). These findings illustrated that the global transcription profiles of the three groups might be not significantly different. Therefore, the difference between baseline (group A) and after entering the simulated high-altitude environment (group B or C) was further explored. Similar to the previous results, although clear between-group differences were observed in the score plots and the models possessed a reasonable fit, the models were overfitting with low predictive performance (Additional file 1: Fig. S1). Additionally, different groups were indistinguishable in the RF models (Fig. 2).
Fig. 1
Plots of the multivariate statistical comparisons among groups based on transcriptional profiles. a The score plot of the principal component analysis (PCA). Each point represents an individual sample. b The PLS–DA score scatter plot. c Permutation test for the PLS–DA model. Abscissa represents the permutation retention of permutation test, ordinate indicates the value of R2 (green dot) and Q2 (blue square) permutation test, and the two dotted lines represent the regression lines of R2 and Q2 respectively. R2 indicates the proportion of variance in the data explained by the model, and Q2 indicates the predictive accuracy of the model. Group A, after adapting to an altitude equivalent to sea level for 1 h. Group B, after adapting to an altitude equivalent to 3500 m for 1 h. Group C, after adapting to an altitude equivalent to 4500 m for 1 h
Fig. 2
Random forest models for the exploration of group differences based on transcriptional profiles. a Multi-dimensional scaling (MDS) plot shows no differentiation among the three groups by random forest model. b MDS plot of random forest model to differentiate groups A and B. c MDS plot of random forest model to differentiate groups A and C
Nevertheless, pairwise differential expression analysis was further performed with the three groups (i.e., comparison between baseline [group A] and after adapting to an altitude equivalent to 3500 m for 1 h [group B]; comparison between baseline [group A] and after adapting to an altitude equivalent to 4500 m for 1 h [group C]) to identify differentially expressed genes (DEGs). A total of 93 differential genes were generated after 1 h post adaptation in a simulated plateau environment with 3500 m altitude, including 67 downregulated and 26 upregulated genes (Fig. 3a). Moreover, a total of 45 differential genes for group C was identified, of which three were expressed upregulated and 42 were expressed downregulated (Fig. 3b). The expression levels of these genes were provided as a heatmap (Additional file 2: Fig. S2a-b). Then, the Boruta algorithm, based on the RF machine learning algorithm, further confirmed seven (comparison between group A and B, Fig. 3c) and eight (comparison between group A and C, Fig. 3d) target genes, respectively, most of which were downregulated genes (Additional file 2: Fig. S2c-d). Moreover, we observed that inter-individual variability of these important genes was low. Taken together, the above results revealed that there were few differences in blood transcriptional profiles before and after entering into the simulated high-altitude environment.
Fig. 3
Differentially expressed genes were identified by differential expression analysis and the Boruta algorithm. a The volcano plot indicates the results of the pairwise comparisons of genes in the blood samples from groups A and B. b Volcano plot presents the differences between groups A and C. c Relative importance of important genes between groups A and B computed using the Boruta algorithm. The horizontal axis is the names of genes, and the vertical axis is the Z-value of each gene. The box plot shows the Z-value of each gene during model calculation. d Relative importance of important genes between groups A and C
ATAC-seq analysis showed less noticeable differences between three groupsAn ATAC-seq analysis was performed to explore changes in accessible regions in the genome after adaption to the simulated high-altitude environment with the three groups. We focused on the peaks located on the promoter region. A PCA plot showed that the three groups could not be distinguished from each other on the basis of PC1 (13.53%) and PC2 (6.56%), which was further confirmed by PERMANOVA analysis (sample: R2 = 0.392, P = 0.001; group: R2 = 0.074, P = 0.482) (Fig. 4a). Different groups were clearly discriminated by the PLS-DA plot; however, these models were overfitting (Fig. 4b, c, Additional file 3: Fig. S3). Similar results were observed in the RF models (Fig. 5). Subsequently, pairwise comparative analysis based on the ATAC-seq data obtained from the three groups were performed to evaluate the differentially accessible regions (DARs), which resulted in 12 (comparison between group A and B, Fig. 6a) and 15 (comparison between group A and C, Fig. 6b) DARs, respectively. We then plotted the expression levels of these DARs as a heatmap (Additional file 4: Fig. S4a-b). Additionally, using the Boruta algorithm, four (comparison between group A and B, Fig. 6c) and nine (comparison between group A and C, Fig. 6d) peaks were identified as important. The chromatin accessibility of the genes related to these important peaks exhibited large heterogeneity between groups (Additional file 4: Fig. S4c-d). These findings illustrated that the ATAC-Seq profiles in blood associated with the promoter region were less impacted by a short period of adaptation to the simulated high-altitude environment.
Fig. 4
Plots of the multivariate statistical comparisons among groups based on the ATAC-seq data. a The score plot of PCA. b The PLS–DA score scatter plot. c Cross-validation plot with a permutation test repeated 200 times
Fig. 5
Random forest models based on the ATAC-seq data. a MDS plot of random forest model to differentiate groups A and B. b MDS plot of random forest model to differentiate groups A and C
Fig. 6
Differentially accessible regions were identified by differential expression analysis and the Boruta algorithm. a Volcano plot showing differential accessible regions between groups A and B. b Volcano plot showing differential accessible regions between groups A and C. c Relative importance of important peaks between groups A and B. d Relative importance of important peaks between groups A and C
Combined analysis of RNA-seq and ATAC-seq screened gene interactionTo establish the relationship between changes in gene expression and chromatin accessibility, we combined the results of RNA-seq with the results of ATAC-seq. A PPI analysis was performed on the 100 DEGs and 16 DARs identified in 3500 m altitude adaption as well as 53 DEGs and 20 DARs identified in 4500 m altitude adaption (Fig. 7a, Fig. 8a). The PPI results indicate interactions between DEGs identified by RNA-seq and DARs identified by ATAC-seq. Additionally, interactions between DEGs and DARs identified through differential expression analysis and the Boruta algorithm were observed. Then, CytoHubba was used to screen top 10 hub genes for each of the 3500 m and 4500 m altitude adaptations (Fig. 7b, Fig. 8b). Among these, CREB-binding protein (CREBBP), tumor necrosis factor receptor-associated protein (TRAP1), tubulin (TUB), and DnaJ (Hsp40) homolog subfamily A member 3 (DNAJA3) were the shared hub genes in adaptation to altitudes in 3500 m and 4500 m.
Fig. 7
PPI network construction and hub gene identification from groups A and B. a PPI network of all DEGs and DARs. b PPI network of the ten hub genes. Different colors represent different sequencing using different screening methods. Different shapes represent different sequencing techniques
Fig. 8
PPI network construction and hub gene selection from groups A and C. a PPI network of all DEGs and DARs. b PPI network of the ten hub genes
In the biological process category, GO enrichment results revealed that hub genes primarily participated in response to hypoxia, response to decreased oxygen levels, response to oxygen levels, response to oxidative stress, and regulation of cellular response to oxidative stress. In the cellular component category, hub genes were primarily associated with the mitochondrial matrix, mitochondrial intermembrane space, histone acetyltransferase complex, and acetyltransferase complex. In terms of molecular function, hub genes were primarily involved in protein folding chaperone, unfolded protein binding, adenosine triphosphate (ATP) hydrolysis activity, histone acetyltransferase activity, ATP-dependent protein folding chaperone, and p53 binding (Fig. 9). KEGG pathway enrichment analysis showed that hub genes were mainly enriched in the viral carcinogenesis, Notch signaling pathway, long-term potentiation, and renal cell carcinoma (Fig. 10). Notably, HIF-1 signaling pathway was enriched in 4500 m altitude adaptations, not in 3500 m altitude adaptations. Accordingly, GO and KEGG enrichment analyses both showed that hub genes were mainly enriched in biological processes and pathways closely related to hypoxia adaptation.
Fig. 9
GO enrichment analysis of hub genes. a GO enrichment analysis between groups A and B. b GO enrichment analysis between groups A and C. The hub genes were enriched into three classifications of biological process (BP), molecular function (MF), and cellular components (CC)
Fig. 10
KEGG enrichment analysis of ten hub genes. a KEGG enrichment analysis between groups A and B. b KEGG enrichment analysis between groups A and C
留言 (0)