
記住我
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in infancy, with an estimated incidence of around 1/28,000–1/50,000 live births (1–4). Over 30 genes have been identified to date as causes of HI, including genes for isolated HI as well as HI associated with multiple genetic syndromes (5). Even so, the genetic cause remains elusive in 21% of people with HI (6). The phenotype of HI is variable, regarding severity, triggers of hypoglycemia, and responsiveness to diazoxide, the only drug with regulatory approval for this indication. There are several histological subtypes of HI, including diffuse, focal and atypical pathology (7, 8). Diffuse HI, due to germline variants in associated genes, affects the entire pancreas and is characterized by β-cell nucleomegaly (9). Focal HI, which is diazoxide-unresponsive (6), results from somatic maternal loss of heterozygosity of chromosome 11p15 coupled with a paternal recessive variant in ABCC8 (OMIM 600509) or KCNJ11 (OMIM 600937) (10), causing an adenomatous hyperplastic lesion within the pancreas (9). Atypical histology, also referred to as Localized Islet Nuclear Enlargement (LINE) or mosaic HI, is associated with diazoxide-unresponsive HI due to mosaic variants in ABCC8 and GCK (OMIM 138079) with histology similar to diffuse HI but restricted to only part of the pancreas (8). Management differs between the different subtypes of HI (11) (Table 1).
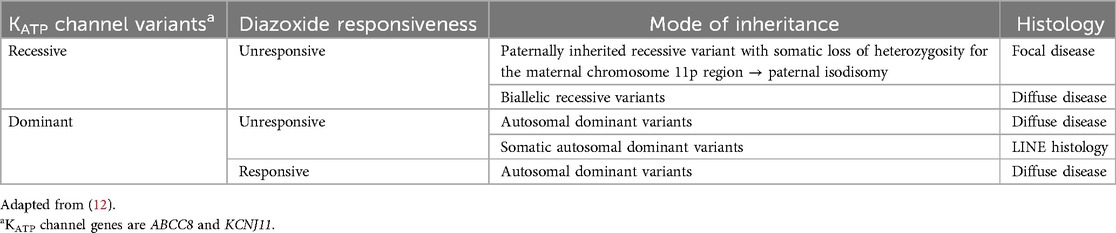
Table 1. KATP channel molecular pathways in hyperinsulinism.
Results of genetic testing can directly inform medical care for patients due to the strong genotype-phenotype correlations for several of the most common causes of HI (6, 11). For example, a paternally inherited ABCC8 or KCNJ11 recessive variant has a 94% positive predictive value for the presence of a focal pancreatic lesion in someone with diazoxide unresponsive HI (6). This result therefore indicates the need for imaging with 18-F-L 3,4-dihydroxyphenylalanine positron emission tomography (18 F-DOPA PET) scan to confirm the presence and location of a lesion, which can then be surgically excised, resulting in a cure when the lesion is completely removed (13, 14). In addition to the implications for management, understanding the genetic etiology also allows accurate genetic counseling to be provided to the family. Depending on the genetic etiology, HI could be due to autosomal recessive, autosomal dominant or rarely X-linked variants (6, 15), in addition to chromosome and imprinting defects (as observed in patients with Turner syndrome or Beckwith Wiedemann syndrome, respectively) (16, 17). While some forms of HI are inherited from affected or unaffected/carrier parents, other genetic alterations occur de novo in the child (6). Because of the complex nature of the potential genetic etiology of a given patient's HI, genetic counseling is an important component of the multidisciplinary care these patients and families require. We present two families affected by HI to illustrate the importance of providing comprehensive genetic counseling to families following a diagnosis of HI.
2 Case reportThe first family (Figure 1) presented at an outside hospital with a newborn male who had persistent hypoglycemia at birth and was ultimately diagnosed with HI. He was initially treated with a glucose infusion rate (GIR) up to 30 mg/kg/min, as well as diazoxide, hydrocortisone and octreotide. Patient was transferred to our Hyperinsulinism Center, and a focal lesion was identified on 18 F-DOPA PET scan. The focal pancreatic lesion was surgically removed following two pancreatectomies (10% pancreatectomy in total). A fasting test proved resolution of the HI prior to discharge. Subsequent genetic testing revealed a paternally inherited recessive ABCC8 variant (c.4480C>T, p.Arg1494Trp, NM_001287174.1). Several years later the paternal half-sister of this patient was diagnosed with HI. She required a GIR of 15 mg/kg/min and was additionally treated with continuous feeds and intravenous glucagon. Genetic testing revealed compound heterozygous pathogenic/likely pathogenic ABCC8 variants [c.4261C>T, p.Arg1421Cys, NM_001287174.1 (maternally inherited); c.4480C>T, p.Arg1494Trp, NM_001287174.1 [(paternally inherited)] and a third ABCC8 variant [c.1063G>A, p.Ala355Thr, NM_001287174.1 (maternally inherited)] classified as likely benign. These results were consistent with diffuse disease. Parents were unaware of the maternal ABCC8 variants prior to evaluation of their daughter. Patient was transferred to our Hyperinsulinism Center for further management and ultimately required a 98% pancreatectomy. Histology revealed scattered islet cell nucleomegaly throughout the pancreas consistent with diffuse disease. Genetic counseling was provided to the family.
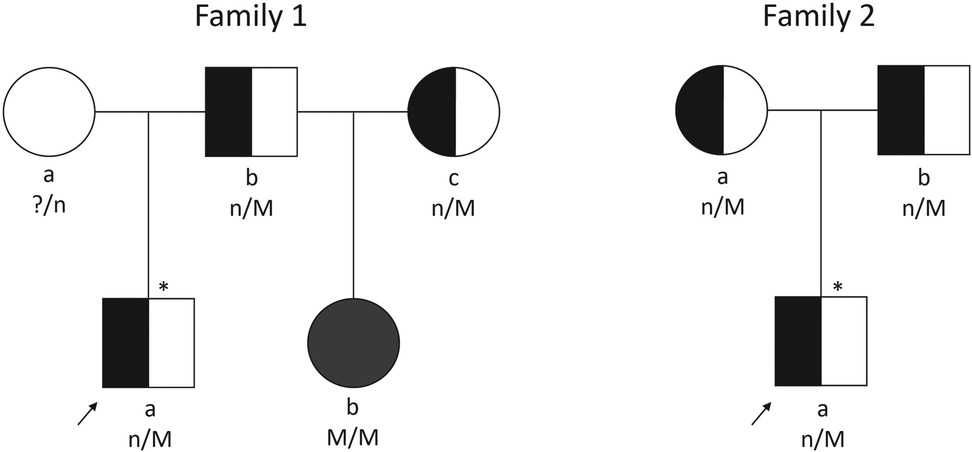
Figure 1. Pedigrees of two families described. Arrows indicate probands. Squares depict males and circles depict females. Filled symbols indicate diffuse HI. Half-shaded symbols indicate carrier of HI; empty symbol indicates unaffected with HI; * indicates focal HI; n/M, mutation carrier; n/n, mutation negative; ?/n, carrier status unknown; M/M, two mutations.
The second family's (Figure 1) 5-month-old son presented to an outside hospital with seizure activity and a plasma glucose of 47 mg/dl (2.6 mmol/L). He was subsequently diagnosed with HI following results of critical laboratory studies and transferred to our Hyperinsulinism Center after failing a diazoxide trial. Genetic testing identified a single recessive ABCC8 variant (c.4013G>A, p.Trp1338*, NM_000352.4). Parental testing revealed both parents carried this variant. 18-F-DOPA PET scan identified a possible exophytic focal lesion arising from the pancreatic tail which was subsequently removed (2% pancreatectomy). Histological exam of the resected lesion noted well-circumscribed proliferation of endocrine tissue with absent p57 immunohistochemical staining of endocrine cell nuclei within the lesion. A cure fast revealed resolving HI prior to discharge. Patient underwent another cure fast around 13 months of age which confirmed resolution of the HI. Genetic counseling was provided to patient's family.
3 DiscussionGenetic counseling is an important component of the care families with HI should receive. In the first family, the father was a known carrier of a recessive ABCC8 variant following the diagnosis of his son with focal HI. Carrier testing for the father's partner(s) should have therefore been discussed (18). Following the daughter's diagnosis of diffuse congenital HI due to compound heterozygous recessive ABCC8 variants, parents indicated they were unaware that future children might be at risk for HI after the first child's diagnosis of focal HI given the rare occurrence of focal HI. The carrier frequency for pathogenic ABCC8 variants in people who are not of Ashkenazi Jewish background is relatively low [approximately 1/125–1/177, based on cited population frequency and ABCC8 gene accounting for approximately 40%–45% of cases of HI (19)]. However, in this case if both partners are carriers, recurrence risk is dramatically altered. It increases from 1/540 for focal HI (20) to 1/4 for diffuse HI, of which this family was unaware.
The second family further illustrates why the mother's genotype should be considered when providing genetic counseling to families with focal HI. In this family, the 1/4 chance that the parents would have a child with diffuse HI was significantly higher than the likelihood they would have a child with focal HI (1/540). Despite the lower probability of focal HI for this couple who were unknowingly both carriers of a recessive ABCC8 variant, their child presented with focal HI. In the absence of parental testing, the family may have assumed a low recurrence risk in future pregnancies as the family in the first case assumed. Indeed, this very scenario was reported by Valayannopoulos et al. (21) in a consanguineous family whose first child had focal HI and only the father had genetic testing for the ABCC8 variant identified in the child. This family had a subsequent child with diffuse HI which was the result of a homozygous variant inherited from each parent. Our family as well as the family reported by Valayannopoulos et al. clearly demonstrate that the presence of focal HI in a child does not exclude the possibility that the child's mother is a carrier of a recessive variant in the same gene. We therefore recommend that mothers of children with focal hyperinsulinism have full gene analysis of the gene associated with focal HI in their child prior to the parents having additional children.
These cases underscore the importance of providing comprehensive genetic counseling to families of children with HI. While the patients referenced had focal or diffuse HI due to pathogenic ABCC8 variants, congenital HI has multiple known monogenic causes as well as associations with multiple genetic syndromes. Understanding the etiology of a patient's HI not only allows for appropriate medical management of the patient (11) but has important reproductive implications for the parents and family. When this increased risk is not known, families may be surprised by having additional medically complex children (21); infants suffering from hypoglycemia may go undiagnosed for many days to months, leaving them vulnerable to the morbidities associated with chronic or severe hypoglycemia, including seizures, developmental delays and cognitive impairments (21–24), and there may be delays in diagnosis (25–27) or in referrals to specialists who care for and treat patients with HI (22). Couples that are aware of their increased risk of congenital HI may defer having additional children or choose from multiple reproductive options including preimplantation genetic testing with in vitro fertilization, prenatal testing via chorionic villi sampling or amniocentesis, use of a gamete donor, adoption, or prompt diagnosis with glucose monitoring and genetic testing at birth. Genetic counselors are uniquely equipped to consider the implications of a genetic diagnosis for the whole family and can assist families and the medical team with the different considerations described above, including carrier testing for the mothers of children with focal HI and discussion of available reproductive options (28). Genetic counseling is therefore an important component of the multidisciplinary care received by every patient and family with HI.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statementWritten informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributionsVS: Conceptualization, Writing – original draft, Writing – review & editing. KL: Investigation, Writing – review & editing. WS: Investigation, Writing – review & editing. HM: Investigation, Writing – review & editing. NA: Investigation, Writing – review & editing. LS: Investigation, Writing – review & editing. TB: Investigation, Writing – review & editing. DDDL: Investigation, Supervision, Writing – review & editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestDDDL has received research funding from Hanmi Pharmaceuticals, Zealand Pharma A/S, Eiger Pharma, Twist Biosciences, Rezolute, Ultragenyx, Moderna, and Crinetics Pharmaceuticals for studies not included in this manuscript. DDDL has received consulting fees from Zealand Pharma A/S, Crinetics Pharmaceuticals, Hanmi Pharmaceuticals, Eiger Pharma, Twist Biosciences, and Rhythm Pharmaceuticals not related to this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Yau D, Laver TW, Dastamani A, Senniappan S, Houghton JAL, Shaikh G, et al. Using referral rates for genetic testing to determine the incidence of a rare disease: the minimal incidence of congenital hyperinsulinism in the UK is 1 in 28,389. PLoS One. (2020) 15(2):e0228417. doi: 10.1371/journal.pone.0228417
PubMed Abstract | Crossref Full Text | Google Scholar
2. Bruining G, Jan MD. Recent advances in hyperinsulinism and the pathogenesis of diabetes mellitus. Curr Opin Pediatr. (1990) 2(4):758–65. doi: 10.1097/00008480-199008000-00024
Crossref Full Text | Google Scholar
3. Otonkoski T, Ammälä C, Huopio H, Cote GJ, Chapman J, Cosgrove K, et al. A point mutation inactivating the sulfonylurea receptor causes the severe form of persistent hyperinsulinemic hypoglycemia of infancy in Finland. Diabetes. (1999) 48(2):408–15. doi: 10.2337/diabetes.48.2.408
PubMed Abstract | Crossref Full Text | Google Scholar
4. Yorifuji T, Horikawa R, Hasegawa T, Adachi M, Soneda S, Minagawa M, et al. (On behalf of the Japanese Society for Pediatric Endocrinology and the Japanese Society of Pediatric Surgeons) clinical practice guidelines for congenital hyperinsulinism. Clin Pediatr Endocrinol. (2017) 26(3):127–52. doi: 10.1297/cpe.26.127
PubMed Abstract | Crossref Full Text | Google Scholar
5. Hewat TI, Johnson MB, Flanagan SE. Congenital hyperinsulinism: current laboratory-based approaches to the genetic diagnosis of a heterogeneous disease. Front Endocrinol (Lausanne). (2022) 13:873254. doi: 10.3389/fendo.2022.873254
PubMed Abstract | Crossref Full Text | Google Scholar
6. Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. (2013) 98(2):E355–63. doi: 10.1210/jc.2012-2169
PubMed Abstract | Crossref Full Text | Google Scholar
7. Sempoux C, Guiot Y, Lefevre A, Nihoul-Fekete C, Jaubert F, Saudubray JM, et al. Neonatal hyperinsulinemic hypoglycemia: heterogeneity of the syndrome and keys for differential diagnosis. J Clin Endocrinol Metab. (1998) 83(5):1455–61. doi: 10.1210/jcem.83.5.4768
PubMed Abstract | Crossref Full Text | Google Scholar
8. Boodhansingh KE, Yang Z, Li C, Chen P, Lord K, Becker SA, et al. Localized islet nuclear enlargement hyperinsulinism (LINE-HI) due to ABCC8 and GCK mosaic mutations. Eur J Endocrinol. (2022) 187(2):301–13. doi: 10.1530/EJE-21-1095
PubMed Abstract | Crossref Full Text | Google Scholar
9. Suchi M, Thornton PS, Adzick NS, MacMullen C, Ganguly A, Stanley CA, et al. Congenital hyperinsulinism: intraoperative biopsy interpretation can direct the extent of pancreatectomy. Am J Surg Pathol. (2004) 28(10):1326–35. doi: 10.1097/01.pas.0000138000.61897.32
PubMed Abstract | Crossref Full Text | Google Scholar
10. Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J Clin Invest. (1998) 102(7):1286–91. doi: 10.1172/JCI4495
PubMed Abstract | Crossref Full Text | Google Scholar
12. Rayannavar A, Christesen HT, De Leon DD. Diazoxide unresponsive forms of congenital hyperinsulinism. In: De Leon-Crutchlow DD, Stanley CA, editors. Congenital Hyperinsulinism: A Practical Guide to Diagnosis and Management. Cham, Humana Press (2019). p. 33–47. doi: 10.1007/978-3-030-02961-6_3
Crossref Full Text | Google Scholar
13. Adzick NS, Thornton PS, Stanley CA, Kaye RD, Ruchelli E. A multidisciplinary approach to the focal form of congenital hyperinsulinism leads to successful treatment by partial pancreatectomy. J Pediatr Surg. (2004) 39(3):270–5. doi: 10.1016/j.jpedsurg.2003.11.019
PubMed Abstract | Crossref Full Text | Google Scholar
14. Hardy OT, Hernandez-Pampaloni M, Saffer JR, Suchi M, Ruchelli E, Zhuang H, et al. Diagnosis and localization of focal congenital hyperinsulinism by 18F-fluorodopa PET scan. J Pediatr. (2007) 150(2):140–5. doi: 10.1016/j.jpeds.2006.08.028
PubMed Abstract | Crossref Full Text | Google Scholar
15. Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, Nelakuditi V, et al. Congenital hyperinsulinism as the presenting feature of Kabuki syndrome: clinical and molecular characterization of 9 affected individuals. Genet Med. (2019) 21(1):233–42. doi: 10.1038/s41436-018-0013-9
PubMed Abstract | Crossref Full Text | Google Scholar
16. Gibson CE, Boodhansingh KE, Li C, Conlin L, Chen P, Becker SA, et al. Congenital hyperinsulinism in infants with turner syndrome: possible association with monosomy X and KDM6A haploinsufficiency. Horm Res Paediatr. (2018) 89(6):413–22. doi: 10.1159/000488347
PubMed Abstract | Crossref Full Text | Google Scholar
17. Kalish JM, Boodhansingh KE, Bhatti TR, Ganguly A, Conlin LK, Becker SA, et al. Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. J Med Genet. (2016) 53(1):53–61. doi: 10.1136/jmedgenet-2015-103394
PubMed Abstract | Crossref Full Text | Google Scholar
18. American College of Obstetricians and Gynecologists' Committee on Genetics. Committee opinion no. 691: carrier screening for genetic conditions. Obstet Gynecol. (2017) 129(3):e41–55. doi: 10.1097/AOG.0000000000001952
PubMed Abstract | Crossref Full Text | Google Scholar
19. Gillis D. Familial hyperinsulinism. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993–2024 (2003). (updated March 21, 2019). p. 1–20.
20. Glaser B, Blech I, Krakinovsky Y, Ekstein J, Gillis D, Mazor-Aronovitch K, et al. ABCC8 mutation allele frequency in the Ashkenazi Jewish population and risk of focal hyperinsulinemic hypoglycemia. Genet Med. (2011) 13(10):891–4. doi: 10.1097/GIM.0b013e31821fea33
PubMed Abstract | Crossref Full Text | Google Scholar
21. Valayannopoulos V, Vaxillaire M, Aigrain Y, Jaubert F, Bellanné-Chantelot C, Ribeiro MJ, et al. Coexistence in the same family of both focal and diffuse forms of hyperinsulinism. Diabetes Care. (2007) 30(6):1590–2. doi: 10.2337/dc06-2327
PubMed Abstract | Crossref Full Text | Google Scholar
22. Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both low blood glucose and insufficient treatment confer risk of neurodevelopmental impairment in congenital hyperinsulinism: a multinational cohort study. Front Endocrinol (Lausanne). (2017) 8:156. doi: 10.3389/fendo.2017.00156
PubMed Abstract | Crossref Full Text | Google Scholar
23. Rother KI, Matsumoto JM, Rasmussen NH, Schwenk WF. Subtotal pancreatectomy for hypoglycemia due to congenital hyperinsulinism: long-term follow-up of neurodevelopmental and pancreatic function. Pediatr Diabetes. (2001) 2(3):115–22. doi: 10.1034/j.1399-5448.2001.002003115.x
PubMed Abstract | Crossref Full Text | Google Scholar
24. Ludwig A, Enke S, Heindorf J, Empting S, Meissner T, Mohnike K. Formal neurocognitive testing in 60 patients with congenital hyperinsulinism. Horm Res Paediatr. (2018) 89(1):1–6. doi: 10.1159/000481774
PubMed Abstract | Crossref Full Text | Google Scholar
25. Hoe FM, Thornton PS, Wanner LA, Steinkrauss L, Simmons RA, Stanley CA. Clinical features and insulin regulation in infants with a syndrome of prolonged neonatal hyperinsulinism. J Pediatr. (2006) 148(2):207–12. doi: 10.1016/j.jpeds.2005.10.002
PubMed Abstract | Crossref Full Text | Google Scholar
26. Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De León DD. High risk of diabetes and neurobehavioral deficits in individuals with surgically treated hyperinsulinism. J Clin Endocrinol Metab. (2015) 100(11):4133–9. doi: 10.1210/jc.2015-2539
PubMed Abstract | Crossref Full Text | Google Scholar
27. Sigal WM, Alzahrani O, Guadalupe GM, Guzman H, Radcliffe J, Thomas NH, et al. Natural history and neurodevelopmental outcomes in perinatal stress induced hyperinsulinism. Front Pediatr. (2022) 10:999274. doi: 10.3389/fped.2022.999274
PubMed Abstract | Crossref Full Text | Google Scholar
28. National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, et al. A new definition of genetic counseling: national society of genetic counselors’ task force report. J Genet Couns. (2006) 15(2):77–83. doi: 10.1007/s10897-005-9014-3
留言 (0)