The three cases presented above highlight the challenging process of establishing the diagnosis and identifying the unique underlying pathology in patients with endocrine hypertension. Further, case management exemplifies the gratifying results of a complete cure of the disease process by appropriate treatment, resulting in desired clinical outcomes. It is crucial for clinicians to have a basic understanding of these disorders for appropriate diagnostic evaluation and to plan for the most desirable treatment on time to avoid catastrophic complications from the underlying disease.
When to Suspect Endocrine Hypertension?
Despite increasing awareness among clinicians, endocrine disorders remain a significant and often neglected cause of SH. The most common causes of endocrine SH are associated with adrenal gland pathology, but more than 15 different disease entities that affect the endocrine system can cause hypertension [4]. SH phenotypes pose a high cardiovascular risk, and the potential curability is the most significant advantage of establishing the underlying cause of hypertension [5, 6]. On the other hand, delayed diagnosis and treatment of secondary hypertension could have deleterious consequences on vital organs. For that reason, clinicians should know when to suspect and actively search for the possible underlying cause of hypertension that will further allow the initiation of adequate therapy. An appropriate case selection is crucial for rationalizing biochemical and hormonal testing and further imaging procedures to avoid unnecessary over-investigations and resource misuse.
Endocrine hypertension may be easily recognized in patients with unique features of hormonal disturbance such as acromegaly, cortisol excess, or pronounced thyroid disorders [7]. However, the symptoms and signs can often be subtle or nonspecific, which can often result in delays in diagnosis. According to the European Societies of Cardiology and Hypertension (ESC/ESH) guidelines, the high-risk hypertensive group of patients in whom the screening for SH is recommended include younger age (below 40 years), acute worsening of BP regulation in previously stable hypertension, severe disease (grade 3 hypertension; BP ≥ 180/120 mm Hg), resistant hypertension, or the presence of extensive hypertension-mediated organ damage [2, 8]. Resistant hypertension is defined as failure to achieve BP below 140/90 mm Hg despite simultaneous use of at least three antihypertensive drugs from different classes, including a diuretic in maximum doses [9]. SH is much more frequent in individuals with resistant hypertension, with a reported prevalence of 31% [10], compared to 15% in the general hypertensive population [2].
The distinctive clinical constellations and typical paroxysms can point to the right diagnosis, as in pheochromocytoma. Furthermore, some laboratory findings (like spontaneous hypokalemia in the absence of any apparent cause) or incidental tumor mass in the suprarenal gland may guide clinicians to investigate the presence of endocrine SH [2].
Primary Aldosteronism
The diagnostic and therapeutic landscape of primary aldosteronism (PA) has changed tremendously since the first reports of the disease, particularly in the last two decades. Initially, PA was considered to be a rare cause of hypertension, accompanied by hypokalemia, periodic muscle weakness, and metabolic alkalosis [11]. From that point, it took a long way to the modern approach, which perceives PA as a syndrome, a spectrum of disorders characterized by inappropriately high, non-suppressible, and renin-independent aldosterone production, with suppressed baseline renin secretion, or the incapability to stimulate renin secretion [12].
Recently, the 2022 WHO Classification of Adrenal Cortical Tumors proposed a classification of morphological substrates for PA: aldosterone-producing adrenal cortical adenoma (APA), diffuse adrenal cortical hyperplasia (APDH), adrenal cortical nodule (APN), adrenal cortical micronodule (APM), multiple APN or APM (MAPN/MAPM) and adrenal cortical carcinoma (APACC) [13].
Most cases of PA are sporadic, with only 1 – 5% of individuals having the genetic basis [14]. The wide variation in the reported prevalence of familial hyperaldosteronism (FH) is due to the selection bias in genetic testing of patients from different series reported from various regions worldwide. Four forms of FH are recognized, with type I (glucocorticoid remediable aldosteronism) due to a hybrid CYP11B1/CYP11B2 gene being the most common [15, 16]. Table 1 provides a brief description of the four forms of FH.
Table 1 Classification of the types of Familial hyperaldosteronism (FH) with common clinical features, genetic defects, type of inheritance and management options2,14. GRA – glucocorticoid remediable aldosteronism, AD – autosomal dominant, PA – primary aldosteronism, MRA – mineralocorticoid receptor antagonist, CCB – calcium channel blockerSince aldosterone is the main regulator of water and electrolyte balance, the connection between abnormal hypersecretion and hypertension is clear. Besides that, the harmful effects of PA extend beyond hypertension and its direct consequences on organs since aldosterone promotes fibrosis, inflammation, and oxidative stress in specific target tissues, with subsequent renal and CV injury [17, 18].
Despite its clinicopathological effects and the associated complications, PA remains largely underdiagnosed, with less than 2% of people among the at-risk populations even tested for the disease [19]. However, several recent publications on PA have helped to raise global awareness. [2, 12, 15, 20] With improved disease recognition coupled with more accurate diagnostic procedures, the care of patients with the disease could improve further in the coming years.
A systematic review and meta-regression analysis by Kayser et al. showed a huge heterogeneity in the prevalence figures among hypertensive patients, depending on the study population: from 3.2% to 12.7% in primary care and from 1% to 29.8% in referral centers [21]. Almost every patient with PA has hypertension [16], while PA is the most common cause of SH [15]. On the other hand, hypokalemia, traditionally used as a hallmark of the disease, can be found only in 9–37% of all cases of PA, mostly in patients with aldosterone-producing adenoma, and it relates to worse CV outcomes [22]. Current understanding of PA expanded a population group in which an active search for the disease is advised. Thus, the high-risk group patients for PA include those with resistant or severe hypertension or hypertension with hypokalemia, adrenal mass, atrial fibrillation, sleep apnea, or a family history of the disease [12].
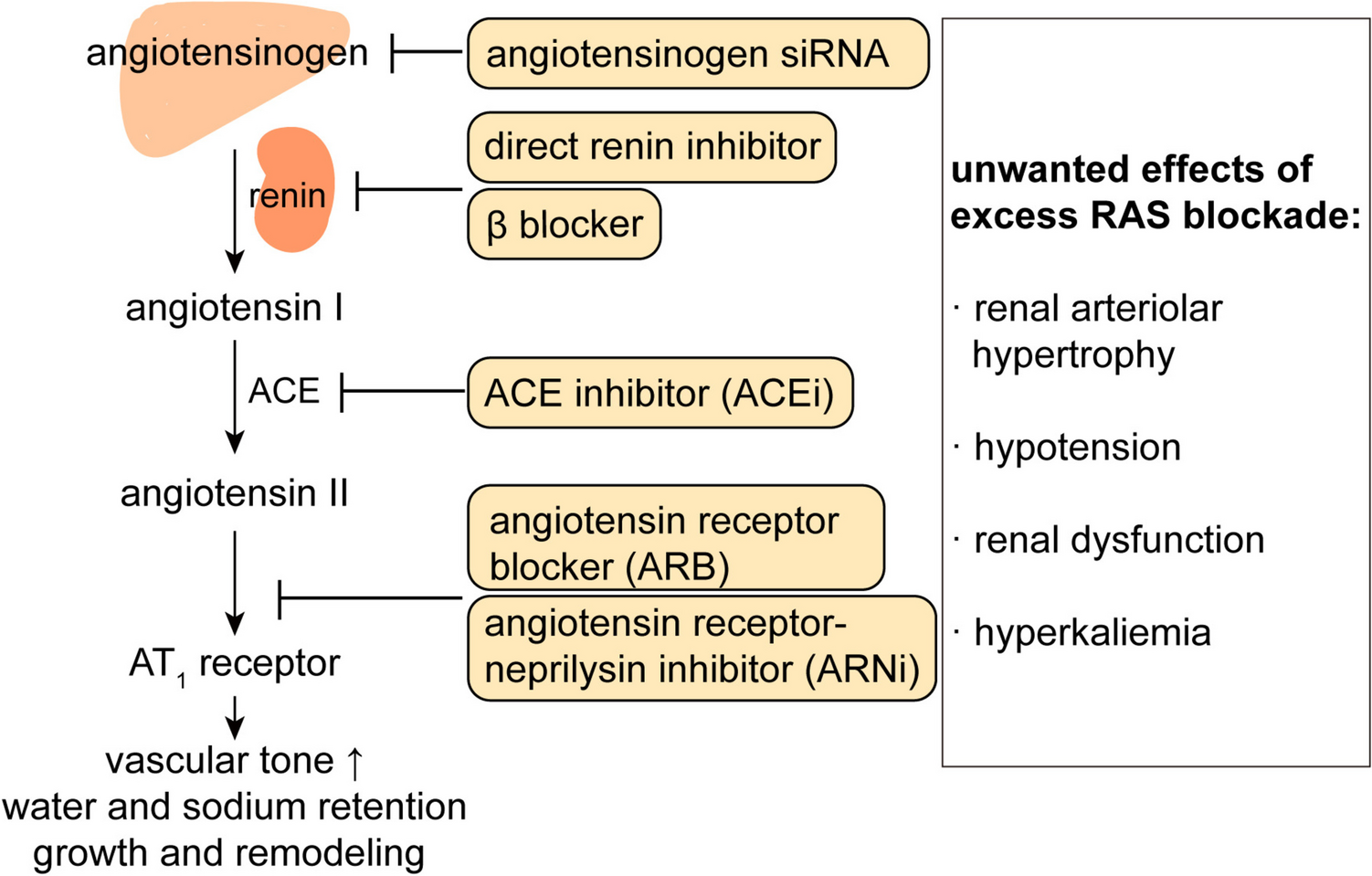
According to the guidelines, the gold standard for case detection as the first step includes the determination of plasma aldosterone to renin ratio (ARR). [15, 23] The potential influence of ongoing antihypertensive drugs on ARR results could be significant. Since, in most cases, as all the antihypertensives cannot be stopped safely, testing may be performed while on treatment. However, the mineralocorticoid receptor antagonists (MRA) must be discontinued for at least 4 weeks. [23] Although different values were suggested, the most widely accepted cut-off to define a positive ARR is 30 when aldosterone is measured in ng/dL and PRA in ng/mL/hr [15].
However, along with the evolving perception of PA as a continuum and data supporting variable ARR performance, some authorities recommend the appraisal of aldosterone and renin values independently (or renin and 24-h urinary aldosterone), not just ARR [12, 24].
Finally, if PAC is between 5 and 15 ng/dl, the patient should undergo confirmatory tests. For this purpose, four dynamic tests in use are the oral sodium loading test, the saline infusion test, the fludrocortisone suppression test, or the captopril challenge test, based on local availability [16, 23], and a non-suppressed aldosterone level suggests autonomous renin-independent overproduction.
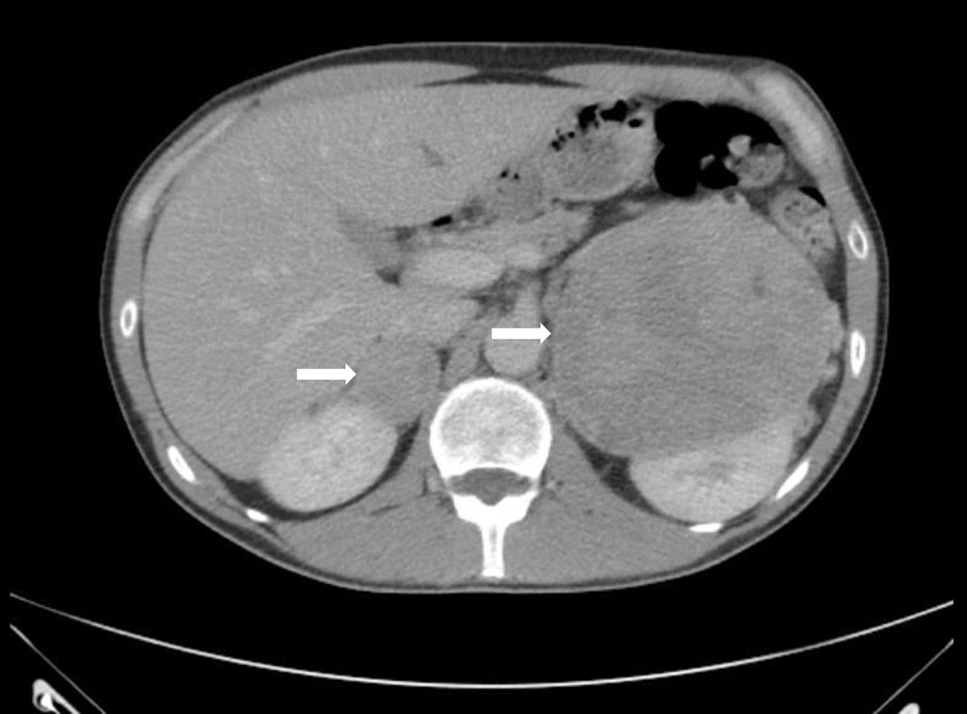
Once the biochemical diagnosis of PA is made, adrenal imaging should be performed. Most widely, a CT scan is used, followed by MRI. As these visualization techniques fail to distinguish between unilateral and bilateral PA, it is recommended to proceed with adrenal vein sampling (AVS) for disease localization to plan definitive treatment, provided the patient agrees to potentially curative surgery. According to an Endocrine Society Clinical Practice Guideline from 2016, in young patients (below 35 years) with spontaneous hypokalemia, severe PAC, and clear unilateral cortical adenoma on imaging, AVS could be skipped before unilateral adrenalectomy [23]. Recently, there has been growing evidence that AVS still should be performed even in such cases [25, 26].
Unilateral forms of PA can be treated surgically, either via laparoscopic or open approach, while bilateral disease is treated by pharmacotherapy using MRAs, such as spironolactone or eplerenone. Surgical treatment can completely cure hypertension in 30–60% of cases and a marked improvement of BP in the remaining patients, leading to a significant reduction in CV risk [27].
Pheochromocytoma & Paragangliomas
Phaeochromocytomas and paragangliomas (PPGL) are rare neuroendocrine tumors with an annual incidence of ≈5 patients per million per year [28]. Despite the low prevalence, raising awareness about these tumors is highly important considering the significant clinical impact due to harmful, potentially life-threatening effects, mostly on the CV system, as timely diagnosis and treatment are crucial to obtain the best outcomes. Genetic mutations are detected in ≈70% of cases [29], and PPGL could be the first manifestation of different inherited syndromes such as von Hippel Lindau (VHL) syndrome, multiple endocrine neoplasia type 2 (MEN2), and neurofibromatosis type 1 (NF1).
Clinicopathological features of PPGL are related primarily to the secretion of adrenaline, noradrenaline, and dopamine in various amounts and proportions. As presented in our case, PPGL can also occasionally co-secrete ACTH, cortisol, calcitonin, vasoactive intestinal peptide, testosterone, renin, aldosterone, and interleukin-6 [30]. Also, a multicenter cross-sectional study conducted by Constantinescu et al., provided insight into a new concept of interactions between the adrenal cortex and medulla [31]. The patients included in their study had higher levels of cortisol, 11-deoxycortisol, 11-deoxycorticosterone, and corticosterone than those with primary hypertension, which certainly contributes to the pathogenesis and severity of hypertension in PPGL.
Pheochromocytomas are diagnosed in 0.2–0.6% of patients with hypertension [32], but up to 95% of patients with pheochromocytomas have high BP, while others are normotensive [32, 33]. Hypertension is considered one of the characteristic features, either episodic or constant. It could be the first manifestation of PPGL, even in the form of a catecholamine-induced hypertensive crisis [34]. High BP is mainly due to vasopressor effects of increased levels of circulating adrenaline and noradrenaline secreted by these tumors [31].
Resistant hypertension and hypertension with typical episodic symptoms of catecholamine excess (headache, palpitations, diaphoresis) are indications for biochemical screening for PPGL. With the widespread use of CT scans and MRI for cross-sectional imaging as a diagnostic evaluation, clinicians often encounter a PPGL when assessing the hormonal activity of adrenal incidentalomas (AI). As per the recently published guidelines, an AI tumor radiodensity of > 10 HU should prompt diagnostic evaluation for a pheochromocytoma [35].
A recently published position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension recommended that the first-line analysis in screening for PPGL should be measurements of plasma or urinary-free normetanephrine and metanephrine with the addition of 3-methoxytyramine in plasma if a dopamine-secreting tumor is suspected [28]. Imaging evaluation with CT scan or MRI is usually sufficient, but in cases with bilateral adrenal masses, additional functional imaging should be performed (positron emission tomography [PET], or 123Iodine meta-iodobenzylguanidine [123I-MIBG] scintigraphy) [16].
The therapeutic approach for PPGL consists of preoperative administration of α-adrenergic receptor blockers (phenoxybenzamine and doxazosin) with subsequent β-blocker administration (with or without other antihypertensive agents) for prompt BP control [36] followed by surgical removal of the tumor (laparoscopic or open approach). When the disease cannot be operated, or with the presence of metastatic disease, the options include chemotherapy, radionuclide ablation procedures, or the use of tyrosine kinase inhibitors [2]. Ideally, all cases should be followed up lifelong with biochemical (and imaging when needed) monitoring to screen for recurrence.
Cushing’s Syndrome
Cushing syndrome (CS), resulting from glucocorticoid excess in the body, is a common sequela of the use of exogenous corticosteroids for various reasons. At times, autonomous overproduction of glucocorticoids can occur in some individuals, resulting in endogenous CS. The pathological hallmark of this type of CS is cortisol excess, either due to adrenocorticotropic hormone (ACTH) overproduction (ACTH-dependent) as in the case of an ACTH-producing pituitary tumor (Cushing disease; CD) or ACTH–independent secretion from an adrenal adenoma. CS, as such, is a rare disease, with an annual incidence of 2 to 8 cases per million population [37]. Rarer forms of CS due to various genetic disorders are also described (beyond the scope of this review and may be referred to elsewhere) [37].
Clinical suspicion for endogenous CS should prompt laboratory evaluation by initial screening tests, including an ODST, 24-h urinary free cortisol (UFC) assay, and/or late-night salivary cortisol (LNSC), depending on local availability [37, 38]. A loss of circadian rhythm with elevated level of cortisol at midnight, high UFC, or failure to suppress cortisol level at 8 a.m. after peroral administration of 1 mg dexamethasone at 11 p.m. the night before requires further biochemical testing and, if CS is confirmed, imaging procedures (CT, MRI, positron emission tomography, bilateral inferior petrosal venous sampling) for tumor localization.
The overall standardized mortality rate (SMR) in Cushing disease (CD) is double than that in the general population. At the same time, coexistent hypertension and diabetes mellitus (DM) are associated with significantly worse survival rates [39]. The leading cause of death is CVD [40, 41], which is of particular concern knowing that patients with cortisol excess have a four- to five-fold higher risk of developing CVD in comparison to the general population [2].
Hypertension is present in approximately 80% of patients with CS [42], independent of their age and sex, and it is multifactorial. It encompasses cortisol binding to the mineralocorticoid receptors with consequent sodium retention and hypervolemia, increased production of endothelin-1 and other vasoconstrictors coupled with inhibition of vasodilator release, but also a modulation of the renin–angiotensin–aldosterone system (RAAS) activity [42,43,44,45].
Since there is a correlation between the duration of uncontrolled hypercortisolemia and the development of hypertension [46], hypertension should be treated as soon as it is diagnosed. Considering the pathophysiological aspects of hypertension in CD, Isidori et al. proposed a tailored treatment algorithm that uses angiotensin-converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB) as the first-line treatment [47]. If the BP control is not achieved, hypokalemia is the next determining factor and if present, spironolactone or eplerenone should be added, and if absent, calcium antagonists are recommended as add-on therapy. In the third line, alpha-blockers, nitric-oxide donors, and cautious use of diuretics or beta-blockers should be considered.
The surgical removal of the cortisol-producing tumor is the primary therapeutic approach in CS patients, but even with remission of hypercortisolemia, hypertension, and other metabolic disturbances do not necessarily normalize and often persist even during the follow-up period [41]. Upon remission of CS, amelioration in cardiac morphology (like hypertrophy) is frequently detected, but structural changes may not be fully resolved [48]. Persistent hypertension during follow-up has been reported in 25–54% of patients in remission [46] and could be an indirect sign of irreversible cardiovascular changes. Pharmacological therapy in CS also has a positive impact on BP values. Pasireotide reduces BP [49], and a similar effect is gained with other drugs that control hypercortisolism [47].
Acromegaly and Growth Hormone Deficiency
As per our current knowledge, both growth hormone (GH) excess and deficiency could be associated with hypertension by different pathobiological mechanisms.
Acromegaly is a rare disease with a prevalence between 2.8 and 13.7 cases per 1,00,000 people [50]. The disease is characterized by GH excess, most commonly originating from a pituitary adenoma, and elevated levels of IGF-1, its primary mediator. Typical clinical features include altered facial appearance (large lips and nose, mandibular overgrowth, prognathism, etc.) and acral enlargement. Acromegaly is strongly associated with CV, metabolic, respiratory, musculoskeletal, neurological, and neoplastic comorbidities, not necessarily completely reversible even after full disease control is achieved [51, 52].
For many years, CVD was the main cause of mortality in patients with acromegaly. Based on raised awareness among clinicians in recent years, a shift to cancer as the leading cause is notable [53]. Hypertension is the most frequent CV manifestation in acromegaly, with an estimated prevalence of about 30% [54]. However, in some cohorts, the prevalence reached 56% at study entry and 64% during the follow-up [55]. Pathogenesis of hypertension is multifactorial, including increased serum GH levels leading to insulin resistance due to the decreased insulin effects on liver and extrahepatic tissues, endothelial dysfunction, increased renal sodium reabsorption, and consequent expansion of extracellular volume, and sleep apnea syndrome [56].
A current therapeutic approach to acromegaly should aim not only for the biochemical normalization of IGF1 levels but also for optimal control of comorbidities and complications [57]. Since hypertension in acromegaly is usually mild, the Acromegaly Consensus Group recommends th




留言 (0)